
 Tiếng Việt
Tiếng Việt| MINISTRY OF HEALTH | SOCIALIST REPUBLIC OF VIETNAM |
| No: 11/2018/TT-BYT | Hanoi, May 4, 2018 |
CIRCULAR
ON DRUG/DRUG INGREDIENT QUALITY
Pursuant to the Law No. 34/2005/QH11 dated June 14, 2005 on Pharmacy;
Pursuant to the Government’s Decree No. 54/2017/ND-CP on detailing a number of articles of, and providing measures for implementing, the Law on Pharmacy dated May 8, 2017;
Pursuant to the Government’s Decree No. 75/2017/ND-CP dated June 20, 2017 defining Functions, Tasks, Powers and Organizational Structure of Ministry of Health;
At the quest of the Director General of the Drug Administration,
The Ministry of Health promulgates the Circular on Drug/Drug Ingredient Quality.
Chapter I
GENERAL PROVISIONS
Article 1. Scope
This Circular provides for application of quality standards of drugs (modern drugs, herbal drugs, vaccines and biological) and drug ingredients (except herbal ones); drug/drug ingredient tests and procedures for recall and handling of unconformable drugs.
Article 2. Definitions
For the purpose of this Circular, the terms below shall be construed as follows:
1. Drug/drug ingredient quality standards are documents regulating technical characteristics of drugs and drug ingredients, including quality criteria, quality levels, test methods and other administrative requirements.
2. GLP stands for Good Laboratory Practice.
3. WHO stands for World Health Organization.
4. ICH stands for International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use.
Chapter II
APPLICATION OF DRUG/DRUG INGREDIENT QUALITY STANDARDS
Article 3. General provisions
1. Pharmacy business establishments and drug preparing facilities shall apply drug/drug ingredient quality standards by way of pharmacopeia or internal standards for drugs and drug ingredients produced and prepared by those facilities.
2. Pharmacy business establishments and drug preparing facilities must carry out evaluations of test methods stated in drug/drug ingredient quality standards published and applied by the drug manufacturers. Assessment and evaluation of test methods are carried out in accordance with guidelines for assessment of analytic processes by ASEAN or ICH, specified in the Circular on Registration of Drugs and Drug Ingredients promulgated by the Minister of Health.
3. The Ministry of Health organizes document assessment and approval of drug/drug ingredient quality standards, in accordance with regulations on drug/drug ingredient registration, issuing permits for drugs /drug ingredients which do not have prior registrations for circulation.
Article 4. Application of pharmacopeia
1. Application of Vietnam’s pharmacopeia and reference pharmacopeias:
a) Pharmacy business establishments and drug preparing facilities can apply Vietnam’s pharmacopeia or one of the following reference pharmacopeias: European, British, United States, International, and Japanese;
b) The application of standards of the pharmacopeias specified in Point a of this Clause must include all regulations on quality criteria, quality levels and test methods specified in the respective drug/drug ingredient’s treatise in the chosen pharmacopeia; also including regulations on quality criteria, quality levels and test methods specified in the appendix of that pharmacopeia;
c) If the manufacturer announces its application of one of the pharmacopeias specified in Point a of this Clause, but adopt test methods different from the ones specified in the drug/drug ingredient’s treatise in the chosen pharmacopeia, the manufacturer must prove their chosen methods' equivalence to the pharmacopeia’s methods. The test results from the methods stated in the pharmacopeia are the basis for evaluation of drug quality;
d) For herbal drugs, pharmacy business establishments and drug preparing facilities can apply the pharmacopeias specified in Point a of this Clause or the pharmacopeia of the drug’s country of origin.
2. Application of pharmacopeias other than the ones specified in Point a of this Clause:
If the pharmacy business establishment or drug preparing facility decides to apply a pharmacopeia other than ones specified in Point a of this Clause, the applied quality standards must at least:
a) Meet the requirements of quality criteria and levels specified in the respective quality criteria’s treatises in Vietnam’s pharmacopeia or one of the aforementioned reference pharmacopeias;
b) The applied common test methods must be appropriate for the equivalent common test methods stated in Vietnam’s pharmacopeia or one of the reference pharmacopeias specified in Point a of this Clause.
Article 5. Application of internal standards
1. The internal drug/drug ingredient standards must conform to the regulations specified in Point b, Clause 2, Article 102 of the Law on Pharmacy, as follows:
a) Meet the requirements of quality criteria and levels specified in the respective treatises in Vietnam’s pharmacopeia and quality criteria, quality levels and common test methods specified in the appendix of Vietnam’s pharmacopeia;
b) If Vietnam's pharmacopeia or the reference pharmacopeias specified in Point a, Clause 1, Article 4 of this Circular do not have any treatise for the required drug/drug ingredient, the facility shall form the standard using the scientific research results (also including the product development research results) or the regulations in other foreign pharmacopeias as the basis.
2. The internal standards of drugs prepared in medical facilities are formed and evaluated for appropriateness by the facility, and promulgated by the facility's head.
Article 6. Update of quality standards and application of updated pharmacopeia
1. When applying for circulation (or circulation extension) of a drug/drug ingredient: The quality standards of that drug/drug ingredient must conform to one of the following pharmacopeias at the time of application:
a) The pharmacopeia’s latest edition;
b) The pharmacopeia’s previous editions which did not come more than two years before the current edition.
2. In the case of drugs or drug ingredients that have already been allowed for circulation: For a maximum of two years from the effective date of the pharmacopeia’s latest edition, the applier or manufacturer have the responsibility to update the standards of drugs/drug ingredients as regulated by that edition.
3. During drug/drug ingredient circulation, if the applier or manufacturer finds any factor that severely affect drug safety, quality or efficacy or is requested by the Ministry of Health (Drug Administration), the manufacturer must the update the drug/drug ingredient standards’ criteria in order to bring that factor under control.
Chapter III
DRUG/DRUG INGREDIENT TESTS
Article 7. Drug/drug ingredient tests
1. The test must be carried out in accordance with the approved and updated drug/drug ingredient quality standards.
If the drug/drug ingredient quality standard is not updated, the testing facility shall use the equivalent pharmacopeia specified in Clauses 1 and 2, Article 6 of this Circular, based on the production date of the drug/drug ingredient being tested.
If the drug is prepared in a medical facility, the test is carried out in accordance with the drug quality standards formed and promulgated by the facility.
2. The collection of drug/drug ingredient samples for testing is carried out in accordance with Appendix I and the sample collection form in Template No. 1 of Appendix III issued together with this Circular.
3. Presenting drug/drug ingredient test/analysis results:
a) The drug/drug ingredient test and analysis results are shown on the test/analysis report based on Samples No. 2 and No. 3 shown in Appendix III issued together with this Circular;
b) The testing facility must present the test/analysis results of the drug sample, which was collected by the quality inspection authority, in no more than 15 days after receiving the drug sample, in the following cases:
- There is information on severely adverse effects of the drug;
- The drug comes from a facility committing serious violations against good practice;
- Additional samples of the drug are collected in the cases mentioned in Point b, Clause 1 and Point b, Clause 2 of this Circular's Article 14.
b) The testing facility must present the test/analysis results of the drug sample in no more than 20 days after receiving it in the following cases:
- The drug requires testing before circulation, as specified in Clause 1, Article 8 of this Circular;
- The drug does not fit any of the cases mentioned in Points b and d of this Clause.
d) The testing facility must present the test/analysis results of the drug/drug ingredient sample in no more than 30 days after receiving it in the following cases:
- The drug/drug ingredient has test methods that require long testing time;
- The drug/drug ingredient requires re-testing or reevaluation of results.
- The drug/drug ingredient has dubious contents or quality, which require test methods other than the ones stated in the registered quality criteria;
- The drug/drug ingredient requires test methods that the testing facility is incapable of conducting (e.g. lack of equipment, chemical, reagents, reference material).
dd) If the deadlines mentioned in Points b, c and d of this Clause are not met, the testing facility has to explain the reason for lateness in a document attached to the test/analysis report;
e) In 24 hours from the time the test/analysis report is issued, the testing facility must send the form to the quality inspection authority, the facilities producing or importing the drug/drug ingredient being tested on and the facility where the sample was taken from.
If the drug/drug ingredient sample does not meet the quality standards, in 24 hours from the time the test/analysis report is issued, the testing facility must notify the Ministry of Health (Drug Administration) of that sample in writing with the test/analysis report attached, both physical and electronic copies (the latter, which is scanned, can be sent to the email address quanlychatluongthuoc.qld@moh.gov.vn or via messaging to the Drug Administration's phone number, with both methods of correspondence using the testing facility's official email address and phone number). A similar notification must also be sent to the Department of Health whose jurisdiction is where the tested drug/drug ingredient comes from.
g) In the case of the drug/drug ingredient sample is sent by a pharmacy business establishment, a facility using it, an organization or an individual for analysis, testing or drug/drug ingredient quality standard assessment, the time of result presentation shall be agreed upon by the parties.
4. Filing and handling of complaints about test results:
a) If there is disagreement with the sample’s test results, in five days from the date the test results are received, the pharmacy business establishment has the right to request the quality inspection authority to assign another testing facility to carry out drug/drug ingredient quality tests/analyses;
b) Re-testing of challenged quality criteria is carried out at the testing facility designated by the Ministry of Health, as specified in Clause 2, Article 105 of the Law on Pharmacy.
5. Retention of samples:
a) The drug/drug ingredient sample must be retained after testing and quality conclusion. The retained sample must be sealed up and preserved as specified in the conditions on the label.
b) Sample retention period:
- In the case of drug/drug ingredient production and importers: the finished product's sample must be retained for a minimum of 12 months after product's expiry date; the active ingredient’s sample must be retained for a minimum of 12 months after the expiry date of the finished product prepared from that ingredient.
- In the case of drug testing facilities: the sample retention period is at least 12 months after the drug’s expiry date, or 24 months after the sample collection date for drug samples collected for quality inspection; or after the date of receipt for additional collected samples specified in Point b, Clause 1 and Point b, Clause 2, Article 14 of this Circular.
6. Archiving records and documents:
a) The records and documents on drug/drug ingredient quality inspection must be archived as specified in the Law on Archives and relevant guiding documents;
b) The records and documents on narcotic, psychiatric, precursor and radioactive drugs/drug ingredients must be archived for a minimum of two years from the expiry dates.
c) After the end of their archive periods, the records and documents shall be handled in accordance with present regulations.
Article 8. Pre-circulation test for drugs specified in Clause 4, Article 103 of the Law on Pharmacy
1. Drugs that belong to one of the following categories must undergo testing carried out by a testing facility designated by the Ministry of Health (Drug Administration) before circulation:
a) The drugs specified in Points a and b, Clause 4, Article 103 of the Law on Pharmacy;
b) Biologicals which are derivatives of human blood and plasma;
c) Imported drugs specified in the Government’s Decree No. 54/2017/ND-CP on detailing a number of articles of, and providing measures for implementing, the Law on Pharmacy dated May 8, 2017;
d) Drugs produced by foreign manufacturers on the list of manufacturers with drugs that do not conform to quality standards, published by the Ministry of Health (Drug Administration).
2. Regulations on drug quality tests:
a) Drug sample collection
- The samples of the drugs specified in Points a, b, c, Clause 1 of this Article shall be collected by manufacturers (in case of domestic drugs) or importers (in case of imported drugs);
- The facilities importing he drugs specified in Point d of this Article shall request the state’s quality inspection or testing authority for to collect samples of those drugs.
b) The importer shall send the drug sample alongside a copy of the producer’s test report to the testing facility specified in Clause 3 of this Article for drug quality inspection in accordance with the approved drug quality standards;
c) Facilities producing or importing vaccines, biologicals which are antisera, derivatives of human blood and plasma mentioned in Points a and b, Clause 1 of this Article shall send the sample as specified in Articles 10 and 11 of this Circular;
d) The testing facility must present the received sample’s test results within the time limit specified in Point c, Clause 3, Article 7 of this Circular.
3. The Ministry of Health (Drug Administration) shall designate a testing facility which is granted certificates of eligibility for pharmacy business including drug testing, or testing facilities specified in Clause 1, Article 35 of the Law of Medicine meeting GLP requirements, to carry out drug tests specified in Clause 1 of this Article.
If the testing facility does not have sufficient capacity for carrying out one or multiple test methods, the testing facility must notify the production/importer and cooperate with the latter in sending samples to other GLP-compliant testing facilities or laboratories compliant to ISO/IEC 17025 which have capacity for carrying out those test methods.
4. The designated testing facility report testing activities to the Ministry of Health (Drug Administration) on a monthly basis, following Template No. 7 of Appendix III issued together with this Circular.
5. The Ministry of Health (Drug Administration) publishes and updates the list of designated testing facilities mentioned in Clause 3 of this Article on the Drug Administration’s website.
6. The production/importer has the responsibility to:
a) Pay its expenses for drug quality tests;
b) Provide reference materials (including those of impurities) to the testing facility if the National Institute of Drug Quality Control, the Institute of Drug Quality Control Ho Chi Minh City, the National Institute for Control of Vaccines and Biologicals or other testing facilities fail to establish;
c) Circulate and distribute the drugs only after their test results show conformity to quality standards.
7. Tests on vaccines, biologicals which are antisera, derivatives of human blood and plasma are carried out in accordance with Articles 10 and 11 of this Circular.
Article 9. The testing periods for facilities on the list of manufacturers with drugs that do not conform to quality standards and withdrawal from that list
1. The testing period starts from the first drug batch's import date after the Ministry of Health (Drug Administration) publishes the list of manufacturers with drugs that do not conform to quality standards and lasts:
a) 6 months for the manufacturer having one drug batch with third-degree violation;
b) 12 months for the manufacturer having one drug batch with second-degree violation or two or more drug batches with third-degree violations;
c) 24 months for the manufacturer having one drug batch with first-degree violation or two or more drug batches with second-degree violations;
d) If the manufacturer continues having uncomformable drugs, the total testing period shall be the sum of individual drugs’ periods.
2. A manufacturer will be withdrawn from the list of manufacturers with drugs that do not conform to quality standards after meeting the following requirements:
a) The manufacturer completes all drug tests before circulation within the time limit specified in Clause 1 of this Article;
The drug manufacturer/registrant files reports which follow Template No. 7 of Appendix III issued together with this Circular, with proof of test on all imported drug batches carried out during the implementation of Clause 1 of this Article;
c) The manufacturer has no drug quality violation (including voluntary drug recall due to quality) during the implementation of Clause 1 of this Article.
3. On a monthly basis, the Ministry of Health (Drug Administration) publishes and updates the list of manufacturers with drugs that do not conform to quality standards, drops the names of facilities complying with the regulations this Article's Clause 2 from the list based on reports from testing facilities that participate in testing activities, drug manufacturers and registrants.
Article 10. Test on vaccines, biologicals which are antisera, derivatives of human blood and plasma
1. The production/importer must send the samples and production records of vaccines, biologicals which are antisera, derivatives of human blood and plasma to the National Institute for Control of Vaccines and Biologicals for testing and evaluation before circulation. The sample sending documents are specified in Article 11 of this Circular.
The production/importer must only circulate vaccines, biologicals which are antisera, derivatives of human blood and plasma after the National Institute for Control of Vaccines and Biologicals confirms the vaccine/biological batches' quality, safety and efficacy and issues quality certificates.
2. Within the time limit specified in Clause 3, Article 7 of this Circular, from the date all samples and documents specified in Article 11 of this Circular are received, National Institute for Control of Vaccines and Biologicals shall:
a) Review the records and conduct tests on the vaccine/biological samples received;
b) Issue quality certificates which follow Template No. 8 of Appendix III issued under this Circular, in which the requirements that are met and which requirements are not, alongside conclusions on the vaccine/biological batch's quality, safety and efficacy;
c) Notify the Ministry of Health (Drug Administration) of the test results.
Article 11. Test samples and records for evaluation of quality, safety and efficacy of vaccines, biologicals which are antisera, derivatives of human blood and plasma
1. For local vaccines, biologicals which are antisera, derivatives of human blood and plasma: The manufacturer shall send the production records and samples from the product batches (either finished semi-finished products) to the National Institute for Control of Vaccines and Biologicals, including:
a) The sample sending form;
b) The vaccine/biological samples to be test on (the number of samples for each kind of vaccine/biological is specified in the Guidelines for testing finished vaccines, biologicals which are antisera, derivatives of human blood and plasma);
c) The records summarizing the vaccine/biological batch’s production and quality tests (copies certified by the manufacturer);
d) The manufacturer’s batch test report.
2. For imported vaccines, biologicals which are antisera, derivatives of human blood and plasma: The importer shall send the production records and samples from the product batches to the National Institute for Control of Vaccines and Biologicals, including:
a) The sample sending form;
b) The vaccine/biological samples to be test on (the number of samples for each kind of vaccine/biological is specified in the Guidelines for testing finished vaccines, biologicals which are antisera, derivatives of human blood and plasma);
c) The records summarizing the imported vaccine/biological batch’s production and quality tests (copies certified by the manufacturer or importer)
d) The quality certificate issued by the country of origin’s authorities for each batch of imported vaccine/biological (copies certified by the importer);
dd) The table of data on preservation conditions (cold storage) during the imported batch’s transport (certified by the importer) from automatic temperature recorders, freeze indicators (if any).
3. The manufacturer and importer must be responsible for their documents' legality.
Chapter IV
REGULATIONS ON RECALL AND HANDLING OF NONCONFORMABLE DRUGS
Article 12. Compulsory drug recall procedure
1. Receiving information on unconformable drugs:
The Ministry of Health (Drug Administration) receives information on unconformable drugs as follows:
a) Information on drugs that do not guarantee effective treatment or is unsafe from the drug registration advisory board or post-vaccination complication handling advisory board;
b) Information on drug quality criteria that are not met from drug testing facilities;
c) Information on discovered unconformable drugs from the Drug Administration, Health/Pharmaceutical inspection authority;
d) Unconformable foreign drug notices from manufacturers, pharmaceutical and drug quality inspection authorities;
dd) Information on unconformable drugs from public security, customs and market surveillance;
e) Drug information from pharmacy business establishments requesting voluntary drug recall.
2. Identification of the violation’s seriousness:
a) In 24 hours from the time the information on unconformable drugs mentioned in Points a, c, d, dd and e, Clause 1 of this Article, the Ministry of Health (Drug Administration) shall identify the violation’s seriousness and draw conclusions on drug recall, based on evaluation of consumer health's risks.
If the drug registration advisory board's opinion is requested for identification of the violation’s seriousness, as specified in Section IV, Appendix II issued together with this Circular, the time limit of identification of violation's seriousness will be 7 days.
b) The seriousness of a drug’s violation is specified in Appendix II issued together with this Circular;
c) For information on unconformable drugs mentioned in Point b, Clause 1 of this Article, the handling shall be carried out in accordance with Article 14 of this Circular.
3. Issuance of drug recall decision:
a) In 24 hours from the time the conclusion on drug recall is drawn, the Ministry of Health (Drug Administration) shall issue the drug recall decision in accordance with Clause 1, Article 65 of the Law on Pharmacy;
b) The drug recall decision must include the following information: drug name, circulation registration number or import permit number, name of active ingredient, concentration, content, form of preparation, batch number, expiry date, manufacturer, importer, recall level, the facility responsible for drug recall.
4. Notification of drug recall decision:
a) The drug recall decision of the Ministry of Health (Drug Administration) is announced by post, fax, email, telephone or the mass media. The scope of drug recall announcement is specified in Clause 3, Article 63 of the Law on Pharmacy;
b) Immediately after making the recall decision, the Ministry of Health (Drug Administration) announces the drug recall decision on websites of the Ministry of Health and the Drug Administration, and the Ministry of Health’s national pharmaceutical database;
Departments of Health announce drug recall decisions on their websites immediately after receiving those decisions.
Domestic drug manufacturers and importers must notify the information about recalled drugs to drug traders/users which purchased those drugs.
c) For recalling drugs with first-degree violations, besides carrying out the actions specified in Point b of this Clause, the Ministry of Health must announce the drug recall decision on Vietnam Television and Voice of Vietnam.
5. Recalling drugs:
a) The drug trader/user must discontinue provision and use of the recalled drugs; place inventory drugs in quarantine; make a list of drug traders/users and individuals (if any) that purchased those drugs, contact them and receive the returned drugs; return the drugs to the providers;
b) The manufacturer (of domestic drugs) and importer cooperate with the import entrustor or distribution hub (of imported drugs) in recalling unconformable drugs. The recall form follows Template No. 4 of Appendix III issued together with this Circular.
The drug trader/provider that fails to recall drugs or receiving returned drugs shall be notified by facilities and individuals purchasing those drugs to the local Department of Health and face actions.
c) Drug recall has to be completed within one of the time limits specified in Clause 3, Article 63 of the Law on Pharmacy.
6. Drug recall report, evaluation and additional measures:
a) In one day (for first-degree recalls) or three days (for second- and third-degree recalls) from the recall’s date of completion, the facility in charge of recalling must report the results to the Ministry of Health (Drug Administration) and the local Department of Health in writing. The report consists of the following documents:
- Summary drug recall report, which follows Template No. 5 of Appendix III issued together with this Circular.
- List of drug traders/users (including those receiving drugs from the facility in charge of recalling unconformable drugs, or from distributors) with their addresses, phone numbers, email addresses (if any), amount of drugs received, amount of drugs recalled;
- Delivery reports, receipts of return or other evidence of drug recall;
- Drug recall self-evaluation form;
- Investigation results, evaluation of causes, evaluation of risks in the unconformable drug's other batches and/or other drugs coming from the same production line.
b) The Ministry of Health (Drug Administration) consider the report mentioned in Point a of this Clause, evaluate it or send it to the Department of Health for evaluation. If the drug recall is evaluated to be insufficient and the product can still be circulated and used, posing a risk to the consumers' health, the Drug Administration cooperates with the Department of Health and other related authorities in coercive drug recall.
Article 13. Voluntary drug recall procedure
1. The pharmacy business establishment that carries out voluntary drug recall shall evaluate and identify the seriousness of the drug's violation and report on the unconformable drug, seriousness of violation, reason for recall and handling measure proposal to the Ministry of Health (Drug Administration) in writing, as specified in Clauses 3 and 4, Article 15 of this Circular.
2. In three days from the date the pharmacy business establishments’ report is received, the Ministry of Health (Drug Administration) consider the report and identify the seriousness of the drug's violation as specified in Appendix II issued together with this Circular.
a) If an agreement with the pharmacy business establishment’s proposal concerning the drug with third-degree violation is reached, the Ministry of Health (Drug Administration) shall send an document allowing the facility to voluntarily recall the drug.
b) In the case of drugs with first- or second-degree violations, the Ministry of Health (Drug Administration) shall follow the drug recall procedures mentioned in Clauses 3, 4, 5 and 6, Article 12 of this Circular;
c) If additional information or clarification of information in the pharmacy business establishment’s report is needed, the Ministry of Health (Drug Administration) shall request the establishment to provide additional information and explanations in writing. In five days from the day the Ministry of Health’s request is received, the establishment must provide additional information and explanations in writing.
3. In 24 hours from the time the Ministry of Health (Drug Administration ) issues the document allowing voluntary drug recall, the establishment can issue the drug recall decision, notify it to traders/users and carry out drug recall as specified in Clauses 5 and 6, Article 12 of this Circular.
Article 14. Handling of drugs not meeting quality standards by place of collection
1. In the case of unconformable drug samples collected from retailers, level-III and level-IV medical facilities:
a) In 24 hours from the time the testing facility’s test/analysis report is received, the Department of Health shall seal the unconformable drug at the sample’s facility of origin.
b) In 48 hours from the time the testing facility’s test/analysis report is received, the Ministry of Health (Drug Administration) shall request the responsible drug registrant/manufacturer/importer to:
- Report its drug distribution to the Ministry of Health (Drug Administration);
- Request the quality inspection authorities to collect additional samples from domestic drug manufacturers or importers, and from at least two wholesalers, with one of them already supplied drugs to the facility where the samples are collected from;
- Send samples to central testing facilities in order to have the unfulfilled criteria tested.
c) If at least one of the additional sample does not meet the quality standards, the Ministry of Health (Drug Administration) shall identify the violation’s seriousness and draw the conclusion on recalling the unconformable drug as specified in Appendix II issued together with this Circular, and issue the drug recall decision as specified in Clause 3, Article 12 of this Circular. The recall's scope and time limit is specified in Clause 3, Article 63 of the Law on Pharmacy;
d) If all of the additional samples meet the quality standards, the Ministry of Health (Drug Administration) shall only carry out the steps of identifying the violation’s seriousness, drawing the conclusion on recalling the unconformable drug, issuing the drug recall decision and drug handling to the drugs of the facility providing the initial samples.
2. In the case of unconformable drug samples collected from wholesalers, level-II or above medical facilities:
a) In 24 hours from the time the testing facility’s test/analysis report is received, the Department of Health shall seal the unconformable drug at the facility of origin.
b) In 48 hours from the time the testing facility’s test/analysis report is received, the Ministry of Health (Drug Administration) shall issue the drug recall decision applying to the province the facility of origin is based on and traders/users receiving the drug from that facility, as specified in Clause 3, Article 12 of this Article, and request the responsible trader/user/importer to:
- Report its drug distribution to the Ministry of Health (Drug Administration);
- Request the quality inspection authorities to collect at least two additional samples from other wholesale establishments, with one of them already supplied drugs to the facility where the samples are collected from;
- Send samples to central testing facilities in order to have the unfulfilled criteria tested.
c) If at least one of the additional sample does not meet the quality standards, the Ministry of Health (Drug Administration) shall identify the violation’s seriousness and draw the conclusion on recalling the unconformable drug as specified in Appendix II issued together with this Circular, and issue the drug recall decision as specified in Clause 3, Article 12 of this Circular. The recall's scope and time limit is specified in Clause 3, Article 63 of the Law on Pharmacy;
d) If all of the additional samples meet the quality standards, the Ministry of Health (Drug Administration) shall only carry out the process specified in Point b of this Clause.
3. If the sample is collected from manufacturers, importers and preservation service providers, or the sample's quality violation is identified to be caused by the production process, the Ministry of Health (Drug Administration) shall identify the violation’s seriousness and draw the conclusion on recalling the unconformable drug as specified in Appendix II issued together with this Circular, and issue the drug recall decision as specified in Clause 3, Article 12 of this Circular. The recall's scope and time limit is specified in Clause 3, Article 63 of the Law on Pharmacy.
Article 15. Handling of recalled drugs
1. The recalled drug can either be rectified or re-exported if it has third-degree violation and does not fall into the type of drug mentioned in Point b, Clause 2 of this Article.
2. The recalled drug must be destroyed if it has:
First- or second-degree violation;
b) Third-degree violation, considered by the Ministry of Health (Drug Administration) to be neither rectifiable nor re-exportable, as specified by Clauses 3 and 4 of this Article;
c) Third-degree violation, considered by the Ministry of Health (Drug Administration) to be rectifiable or re-exportable, but the facility fails to rectify or re-export that drug.
3. Procedure for proposing rectification of recalled drugs:
a) The facility that has recalled drugs shall send the Ministry of Health (Drug Administration) a document stating the rectification process, drug quality and stability risk assessment, the program for monitoring and surveillance of the drug’s quality, safety and efficacy during circulation.
b) In 60 days from the date the facility’s rectification proposal is received, the Ministry of Health (Drug Administration) must consider the proposal and reply their agreement or disagreement in writing The reason for disagreement must be specified;
c) If additional information or clarification of the rectification's information is required, in 60 days from the date the Ministry of Health’s (Drug Administration) document is received, the facility must provide documents additional information and explanations. Failure to do so within the aforementioned time limit will result in invalidation of the rectification proposal.
4. Procedure for proposing re-export of recalled drugs:
a) The facility that has recalled drugs shall send the Ministry of Health (Drug Administration) a document with the re-export plan, stating the time and re-export country;
b) In 15 days from the date the facility’s proposal is received, the Ministry of Health (Drug Administration) shall reply their agreement or disagreement on the re-export in writing; the reason for disagreement must be specified.
5. The rectification and re-export of recalled drug shall only be carried out after the written agreement of the Ministry of Health (Drug Administration) is issued.
6. Drug destruction:
a) The head of the facility that has drugs to be destroyed shall decide to form the drug destruction council. The council shall have at least three persons, with one representative having professional responsibility;
b) Drug destruction must be safe for both humans and animals, does not pollute the environment in accordance with the rules of law in environmental protection;
c) Drug destruction that requires special control must be carried out as specified in Article 48 of Decree No. 54/2017/ND-CP ;
d) The facility carrying out drug destruction must notify the Department of Health, and send the Department a drug destruction form that follows Template No. 6 in Appendix III issued together with this Circular.
7. The recalled drug handling period shall not exceed 12 months from the recall’s date of completion, as specified in Points a, b and c, Clause 3, Article 63 of the Law on Pharmacy.
Article 16. Responsibilities for drug recall
1. Responsibilities of pharmacy business establishments, medical facilities and drug users:
a) Comply with the regulations in Clauses 1, 2 and 3, Article 64 of the Law on Pharmacy;
b) Regularly review and update information on drug recall from websites of the Ministry of Health, the Drug Administration, and Departments of Health.
2. Responsibilities of the Drug Administration:
a) Receive information, identify seriousness of drug’s violations and issue drug recall decisions;
b) Announce drug recall decisions as specified in Point a, Clause 4, Article 12 of this Circular, publish information about recalled drugs on websites of the Ministry of Health and the Drug Administration after those decisions are issued. Cooperate with Vietnam Television and Voice of Vietnam in announcing recall of drugs with first-degree violations;
c) Consider the evaluation reports and reply to the pharmacy business establishments’ proposals for voluntary drug recall, rectification or re-export of recalled drugs;
d) Cooperate with related units (Ministerial Inspector, Department of Health, health divisions of other agencies) in inspection of organization and execution of drug recall; take actions against violating facilities in accordance with the regulations of law;
dd) Produce documents providing detailed guidelines for the processes of drug recall and handling, evaluation of drug recall in drug manufacturers and pharmacy business establishments.
3. Responsibilities of Departments of Health:
a) Publish drug recall decisions on websites of the Departments of Health;
b) Organize announcement and dissemination of drug recall information to local drug manufacturers, pharmacy business establishments and medical facilities;
c) Cooperate with facilities having drugs with quality violations in collecting additional drug samples as specified in Point b, Clause 1 or Point b, Clause 2, Article 14 of this Circular, or direct the testing facilities to do so;
d) Organize surveillance of drug recall in the Departments’ jurisdictions; take actions against and penalize facilities violating drug recall regulations within their competence;
dd) Participate in or carry out evaluations of pharmacy business establishments’ drug recall in the Departments’ jurisdictions, under the Ministry of Health’s (Drug Administration’s) direction. Report any drug manufacturer, importer, wholesalers which are distribution hubs that fail to, or insufficiently, recall drugs to the Ministry of Health (Drug Administration)
e) Organize and participate in coercive drug recall.
Chapter V
IMPLEMENTATION PROVISIONS
Article 17. Effect
1. This Circular is in effect from June 20, 2018.
2. The following documents shall be annulled on the date this Circular comes into effect:
a) The Minister of Health’s Circular No. 09/2010/TT-BYT dated April 28, 2010 providing guidance on drug quality management;
b) The Minister of Health’s Circular No. 04/2010/TT-BYT dated February 12, 2010 providing guidance on sample collection for quality identification.
Article 18. Implementation
1. The Drug Administration has the responsibility to:
a) Preside over and cooperate with related units in organizing propagation, dissemination and implementation of this Circular;
b) Preside over and cooperate with the National Institute of Drug Quality Control, the Institute of Drug Quality Control Ho Chi Minh City, the National Institute for Control of Vaccines and Biologicals in formulating plans to collect drug samples for quality inspection and present those plans to the Ministry of Health for consideration, approval and allocate budget for plan implementation within the Ministry’s competence.
Collect drug samples for quality inspection and update the Ministry of Health’s drug quality inspection database with information on collected drug/drug ingredient samples (including: name of drug/drug ingredient, concentration, content, type of preparation, batch number, expiry date, circulation registration number or import permit number, manufacturer, importer, sample collector) and the drug/drug ingredient’s quality inspection results;
c) Provide scientific and technical information on ensuring drug/drug ingredient quality.
Provide the National Institute of Drug Quality Control and the Institute of Drug Quality Control Ho Chi Minh City with label templates and the quality standard of the drug/drug ingredient that is issued circulation registration certificate or import permit (the updated standard if any changes occur). In the case of vaccines and biological, the label template and quality standard shall be sent to the National Institute for Control of Vaccines and Biologicals;
d) Organize quality inspections on drug/drug ingredients manufactured, prepared, circulated and used nationwide. Direct and survey the drug testing system nationwide. Draw conclusions on drug quality, based on the test results from state-owned drug testing facilities’ and relevant records;
dd) Preside over or participate in carrying out state inspections, inspect and take action against violations against the law in drug quality within the Administration's competence.
2. Departments of Health have the responsibility to:
a) Organize drug quality inspections within their jurisdictions and take actions against violations in accordance with the law;
b) Formulate plans to collect drug samples for quality inspection and present those plans to the provincial People’s Committees for consideration, approval and allocate budget for plan implementation within the Committees’ competence;
c) Update the Ministry of Health’s drug quality inspection database with information on collected drug/drug ingredient samples (including: name of drug/drug ingredient, concentration, content, type of preparation, batch number, expiry date, circulation registration number or import permit number, manufacturer, importer, sample collector) and the drug/drug ingredient’s quality inspection results.
3. Responsibilities of the drug testing system:
a) Central drug testing facilities (National Institute of Drug Quality Control, Institute of Drug Quality Control Ho Chi Minh City, National Institute for Control of Vaccines and Biologicals):
- Analyze and test samples to identify the quality of manufactured, circulated and used drugs/drug ingredients; report the test results to the Ministry of Health (Drug Administration) and the local Department of Health;
- Research, establish and publish on websites of the institute and the Drug Administration the list of reference materials (including those of impurities) for analyses and tests on manufactured, imported, circulated and used in Vietnam;
- The National Institute of Drug Quality Control and the Institute of Drug Quality Control Ho Chi Minh City have the responsibility to provide drug testing centers in assigned provinces with physical and electronic copies of drug/drug ingredient quality standards;
- The National Institute for Control of Vaccines and Biologicals, on an annual basis, review and evaluate vaccine/biological quality trends and present the evaluation to the Ministry of Health for consideration, and formation of guidelines for testing finished vaccines, biologicals which are antisera, derivatives of human blood and plasma (including reviewing vaccine/biological batch quality certificate’s test criteria).
Update information about quality certificate issuance for vaccines, biologicals which are antisera, derivatives of human blood and plasma on websites of the institute and the Drug Administration.
b) Provincial testing facilities:
- Analyze and test samples to identify the quality of manufactured, circulated and used drugs/drug ingredients;
- Report the test results to the Department of Health and the Ministry of Health (Drug Administration).
4. Traders have the responsibility to:
a) Organize researches and carry out implementation of the regulations of law on drug/drug ingredient quality promulgated by this Circular;
b) Implement regulations on inspection, control of drugs/drug ingredients’ source and quality. Carry out quality control in order to ensure drug/drug ingredient quality throughout the facility's operation;
c) Establish a system of records and documents in order to monitor circulation of drugs/drug ingredients. Carry out monitoring and surveillance of the quality of drugs/drug ingredients produced by the facility; timely discover and handle unconformable drugs, report those drugs to the pharmaceutical and drug quality inspection authorities.
5. When the drug quality inspection force has not yet been established at all levels, the Ministry of Health shall assign:
a) The National Institute of Drug Quality Control, the Institute of Drug Quality Control Ho Chi Minh City, the National Institute for Control of Vaccines and Biologicals, by their functions, tasks and jurisdictions, to:
- Formulate plans to collect drug samples for testing and surveillance of drug/drug ingredient quality; reserve, receive and use the annual budget for sample collection and tests on drug/drug ingredient samples;
- Collect drug/drug ingredient samples in accordance with the approved plans at establishments doing pharmacy business and using drugs;
- Update the Ministry of Health’s drug quality inspection database with information on drug/drug ingredient samples collected for quality inspection and those samples’ test results;
- Report the test results to the Ministry of Health (Drug Administration) and the local Department of Health if the drug/drug ingredient samples do not meet the quality standards as specified in Clause 3, Article 7 of this Circular.
- The National Institute of Drug Quality Control shall the drug quality inspection database for the Ministry of Health;
b) Provincial testing facilities:
- Formulate plans to collect drug samples for testing and surveillance of drug/drug ingredient quality; reserve, receive and use the annual budget for sample collection and tests on drug/drug ingredient samples;
- Collect drug/drug ingredient samples for quality inspection in accordance with the approved plans at establishments doing pharmacy business and using drugs;
- Update the Ministry of Health’s drug quality inspection database with information on drug/drug ingredient samples collected for quality inspection and those samples’ test results;
- Report the test results to the Ministry of Health (Drug Administration) and the Department of Health if the drug/drug ingredient samples do not meet the quality standards as specified in Clause 3, Article 7 of this Circular.
Article 19. Implementation responsibilities
The Director General of the Drug Administration, Chief of the Ministry Office, Chief Ministerial Inspector, heads of units affiliated with the Ministry of Health, provincial Departments of Health, pharmacy business establishments, other related authorities, organizations and individuals have the responsibility to implement this Circular.
If any complication arises during implementation, the authorities, organizations and individuals are advised to notify the Ministry of Health (Drug Administration) for consideration and solution.
|
| ON BEHALF OF THE MINISTER |
APPENDIX I
GUIDELINES FOR SAMPLING OF PHARMACEUTICAL PRODUCTS AND PHARMACEUTICAL STARTING MATERIALS FOR QUALITY VERIFICATION
I. Sampling procedures and sampling operations
1. Sampling tools
All sampling tools and implements should be made of inert and clean materials, which should be suitable for each sample type, ensure no effect on sample quality, prevent impurities that cause contamination of samples or cross-contamination and ensure safety of sampler (see Section III)
2. Quantity of sample taken
2.1. The quantity of sample taken for analytical and retention purposes should be calculated according to inspection requirements, pharmaceutical product quality standards, applied pharmaceutical starting materials and testing methods but should be sufficient to allow for at least three analyses or to perform tests to obtain accurate and reliable results.
2.2. Two samples are usually taken from each consignment (one for analytical purpose and one for retention purpose). Where necessary, the number of analytical samples and retention samples may be more than two to be sufficient for testing and retention at relevant organizations.
3. Sampling operations
3.1. Sampling principles:
- Depending on the inspection purpose and each type of product, the sampler should decide to adopt an appropriate sampling method.
- The sampling process should be appropriately supervised and documented. Any signs of non-uniformity or deterioration of pharmaceutical products and containers should be documented.
- The sampling procedure should be such that non-uniformity of pharmaceutical products in each sampling unit and entire consignment can be detected. Signs of non-uniformity include differences in shape, size or colour of particles in crystalline, granular or powdered solid substances; moist crusts on hygroscopic substances; deposits of solid pharmaceutical product in liquid or semi-liquid products; and stratification of liquid products.
- Pooling of the samples from the different portions should be avoided, because this can mask contamination, low potency or other quality problems. Separate samples should be formed from these portions and containers.
- For finished drug products, the sampling procedure should take account of the official and non-official tests required for the individual dosage form (e.g. tablets or parenteral preparations). Non-official tests could include testing for adulteration and counterfeiting.
- It is not recommended to mix the pharmaceutical product removed from a container directly with the one left in that container.
3.2. Sampling procedures
- Carry out physical inspection of the consignment: segregate containers by each type of product and consignment, segregate containers that show any signs of deterioration and do not ensure cleanliness for inspection or sampling. Reject unlabelled sampling units.
- Take sampling units from the consignment of products, open containers to take original samples and reseal the containers from which the samples were taken. Ensure that the quantity of materials in the original samples is sufficient to prepare next samples.
- Gently mix original samples into separate samples of each sampling unit.
- Gently mix separate samples into a common sample.
- Form final samples: take equal fractions from the common sample to form final samples, including analytical samples and retention samples.
3.3. Analytical samples and retention samples should be placed in sealed and labeled containers. A sample container should be labeled with name of the pharmaceutical product, name of the manufacturer, lot number, expiry date, number of containers from which samples were taken, place of sampling, number of samples taken (if samples taken are pharmaceutical starting materials for manufacture of narcotic drugs and psychotropic drugs, precursors used as pharmaceutical products and starting materials for manufacture of radiopharmaceuticals), date of sampling and storage conditions in accordance with the sampling record.
3.4. After the sampling is done, analytical samples and retention samples should be separately sealed to ensure their safety during transportation. The seal should clearly specify the date of sampling and bear at least signatures of the sampler and the representative of the establishment where the sample was taken.
Where necessary, the remainder of the sampling interval should be also sealed to prevent tampering of pharmaceutical products and pharmaceutical starting materials.
3.5. Make a sampling record: The sampling record should contain the batch number, date and place of sampling, storage conditions, notes on possible abnormalities, any other relevant observations and at least the name and signature of the sampler and representative of the establishment where the sample was taken.
In the cases where the quality inspectorate takes samples, the record is required to bear the signature of the inspectorate's head.
In case the representative of the establishment where the sample was taken fails to sign the record, the record should bear the signatures of the sampler and witness.
The record should be made into three copies, which are kept at the establishment where the sample was taken, testing authority and pharmaceutical product quality inspecting authority respectively.
4. Sampling of pharmaceutical starting materials
4.1. In case the material is placed in one container only:
a) Take samples of the solid material: take original samples in different locations of the container (at the top, the bottom or in the middle). If the original samples do not show any visual signs, gently mix them into separate samples.
b) Take samples of the liquid or semi-solid material: if the material is non-homogeneous, gently mix it before sampling.
For example, a stratified liquid may be stirred or a solid deposit in a liquid may be dissolved by gentle warming and stirring.
4.2. In case the consignment of material is placed in multiple containers:
Depending on the sampling purpose, uniformity and quality of the consignment of material, adopt an appropriate sampling method according to Section Clause 9 of this Appendix.
5. Sampling of unpackaged semi-finished products
These products include powdered pharmaceutical products, solutions, syrups, ointments, granules, tablets, injections, etc. that are transported in large containers to the packaging facility. Samples shall be taken from each consignment as follows:
1. If the consignment of products is contained in 1 - 2 containers, open the two containers. If the consignment of products is contained in more than 3 containers, open the three containers. Take at least 3 original samples in different locations of each container.
2. Mix original samples into a common sample, and then form final samples, including analytical and retention samples.
6. Sampling of packaging materials
Samples of packaging materials shall be taken as prescribed in Section 1 Clause 9 of this Appendix.
7. Sampling of finished pharmaceutical products
7.1. Take samples of finished pharmaceutical products to inspect or control quality:
a) Samples should be taken at random and at different locations of the consignment.
b) According to the pharmaceutical product quality standard, the quantity of pharmaceutical product taken should be sufficient to allow for testing and retention. If information is insufficient for accurate calculation of the quantity of pharmaceutical product to be taken, consider the minimum quantity of finished pharmaceutical products provided in Section V of this Appendix.
c) Sampling procedures should be completed according to the guidelines provided in Section II of this Appendix.
7.2. Take samples to carry out visual inspection upon import of pharmaceutical products: the quantity of sample taken to carry out visual inspection is specified in Section IV of this Appendix.
8. Sampling of herbal pharmaceutical starting materials
If herbal medicinal products or partially processed herbal medicinal products, including animals and plants (dried medicinal plants and parts thereof) and minerals are regarded as homogeneous, samples thereof shall be taken as prescribed in Section I Clause 9 “r plan” of this Appendix.
9. Sampling plans for pharmaceutical starting materials and packaging materials
9.1. Before sampling, the sampler should check the integrity and deterioration of the container, and uniformity of products in each sampling unit.
9.2. Sampling should be carried out according to one of the three sampling plans provided in Table 1 below.
Table 1: Values of n, p or r for the N sampling units
| Value of n, p, r | Values of N | ||
| n plan | p plan | r plan | |
| 2 | up to 3 | up to 25 | up to 2 |
| 3 | 4 - 6 | 25 - 56 | 3 - 4 |
| 4 | 7 - 13 | 57 - 100 | 5 - 7 |
| 5 | 14 - 20 | 101 - 156 | 8 - 11 |
| 6 | 21 - 30 | 157 - 225 | 12 - 16 |
| 7 | 31 - 42 |
| 17 - 22 |
| 8 | 43 - 56 |
| 23 - 28 |
| 9 | 57 - 72 |
| 29 - 36 |
| 10 | 73 - 90 |
| 37 - 44 |
a) The n plan
The “n plan” should be used only when the material to be sampled is considered uniform and is supplied from a recognized source. Samples can be withdrawn from any part of the container (usually from the top layer). The “n plan” is based on the formula n = 1 + ![]() , where N is the number of sampling units in the consignment. The value of n is obtained by simple rounding. Original samples are taken from n sampling units selected at random and these are subsequently placed in separate sample containers. These original samples are visually inspected and tested for identity. If the results are concordant, the original samples are combined into a final, common sample from which an analytical sample is prepared, the remainder being kept as a retention sample.
, where N is the number of sampling units in the consignment. The value of n is obtained by simple rounding. Original samples are taken from n sampling units selected at random and these are subsequently placed in separate sample containers. These original samples are visually inspected and tested for identity. If the results are concordant, the original samples are combined into a final, common sample from which an analytical sample is prepared, the remainder being kept as a retention sample.
b) The p plan
The “p plan” may be used when the material is uniform, is received from a recognized source and the main purpose is to test for identity. The p plan is based on the formula p = 0.4![]() , where N is the number of sampling units. The figures for p are obtained by rounding up to the next highest integer. Original samples are taken from each of the N sampling units of the consignment and placed in separate sample containers. These original samples are visually inspected and tested for identity. If the results are concordant, p common samples are formed by appropriate pooling of the original samples (if necessary).
, where N is the number of sampling units. The figures for p are obtained by rounding up to the next highest integer. Original samples are taken from each of the N sampling units of the consignment and placed in separate sample containers. These original samples are visually inspected and tested for identity. If the results are concordant, p common samples are formed by appropriate pooling of the original samples (if necessary).
c) The r plan
The “r plan” may be used when the material is suspected to be non-uniform and/or is received from a source that is not well known, herbal medicinal products or partially processed herbal medicinal products. This plan is based on the formula r = 1.5![]() , where N is the number of sampling units. The figures for r are obtained by rounding up to the next highest integer.
, where N is the number of sampling units. The figures for r are obtained by rounding up to the next highest integer.
Original samples are taken from each of the N sampling units of the consignment and placed in separate sample containers. These original samples are visually inspected and tested for identity. If the results are concordant, r samples are randomly selected and individually subjected to testing. If these results are concordant, the r samples are combined for the retention sample.
9.3. The abovementioned sampling plans are not recommended for sampling of starting materials for identification tests. The GMP-WHO rules shall apply instead.
II. Sampling steps
1. Bulk liquid products
The steps to be considered when sampling bulk liquid products are as follows:
- Read and understand the precautions to be observed for the safe handling of the material.
- Gather together the required sampling equipment (sampling tube or weighted sampling can, sample bottles and labels) and check that all the required items are clean.
- Locate the batch.
- Examine the container(s) for signs of contamination of the batch. Record any faults.
- Examine the labels for obvious differences and signs of changes including obliterations and mislabelling. Record any faults.
- Investigate and clarify the sources of and reasons for any faults before proceeding.
- Choose a liquid-sampling tube of size and orifice suitable for the viscosity of the liquid being sampled.
- Sample the liquid, suspension or emulsion (well stirred, if appropriate) by slowly pushing the open sampling tube vertically down- wards through the liquid so that material is collected from each layer.
- Seal the tube, withdraw it from the bulk liquid, and allow liquid adhering to the outside of the tube to drain. Transfer all the contents of the tube to a clean, labelled sample bottle.
- Repeat steps 8 and 9 until sufficient samples for analytical and retention purposes have been obtained.
- Seal the sample bottle.
- Reseal the container from which the samples were taken and label as “sampled”.
- Clean and dry the sampling tube, observing the relevant safety precautions.
- Sample other required containers in the same manner following steps 8–12 above.
- Clean the sampling tube using the recommended cleaning procedure.
- Deliver the analytical samples to the laboratory and the reserve samples to the retention sample store. Report any aspects of the sampling that should be brought to the attention of the analyst or the inspector.
- Check supplier certificate versus the specifications, if applicable.
2. Powdered starting material
The steps to be considered in sampling a powdered starting material are as follows:
- Read and understand the precautions to be observed for the safe handling of the material.
- Gather together the required sampling equipment (sampling spear, sample bottles and labels) and check that all items are clean.
- Locate the consignment and count the number of containers.
- Examine all the containers for obvious differences and signs of damage. Record any faults.
- Examine all the labels for obvious differences and signs of changes, including obliterations and mislabelling. Record any faults.
- Segregate any damaged containers and those with suspected spoiled contents for separate examination. These should then be referred or rejected and dealt with accordingly.
- Segregate any containers with different batch numbers and treat these separately.
- Number the remaining containers.
- Choose the appropriate sampling plan (n, p or r).
- Choose the containers to be sampled in accordance with the requirements of the chosen plan (by the use of random number tables, by drawing lots or by the use of a random number generator if applicable).
- Open the containers one at a time and inspect the contents. Record any differences.
- Choose a suitable, clean sampling spear and plunge this (gates closed) into the powder so that the point of the spear reaches the bottom of the container.
- Open the gates to allow the powder to enter the spear cavities, then reclose them.
- Withdraw the spear from the container and transfer the spear contents to a labelled sample bottle.
- Repeat steps 12–14 until sufficient material has been collected for analytical and retention requirements.
- Seal the sample bottle.
- Reseal the container from which the samples were withdrawn and label as “sampled”.
- Wipe clean the sampling spear if required, observing the safety precautions, before sampling the other chosen containers.
- Repeat steps 12–18 for each chosen container.
- Clean the sampling spear using the recommended cleaning procedure.
- Deliver the analytical samples to the laboratory and the reserve samples to the retention sample store. Report any aspects of the sampling that should be brought to the attention of the analyst or the inspector.
- Check supplier certificate versus the specifications, if applicable.
3. Packaging materials
The steps to be considered in sampling packaging materials are as follows:
- Check the consignment against any associated documentation.
- Check transit containers for the following and report any deviations as necessary:
+ Correct identification;
+ integrity of seal, if appropriate; and
+ Absence of physical damage.
- Obtain the required sample from the required number of containers, bearing in mind the special considerations for sampling packaging materials noted in Section I Clause 9 of this Appendix.
- Place the sample units into identified appropriate sample containers.
- Identify the consignment containers that have been sampled.
- Note any special situations found during the sampling process (e.g. rogue items or component damage). Report any such observations as necessary.
- Remove all sampled material pallets or containers from the sampling area together with all documentation.
- Check supplier certificate versus the specifications, if applicable.
4. Finished products
The following steps should be considered when sampling finished products:
- Determine the number of pallets per batch in the consignment.
- Calculate the number of pallets according to the number of sampling units to carry out visual inspection:
+ Check condition of pallet and packaging for integrity of outer packaging material.
+ Check outside of goods on the pallets for general cleanliness.
+ Check that the overall labelling of the pallets matches the packing list.
+ Count, categorize and record the number of defects.
- Count the total number of transport packs on the number of pallets present and verify the total against the packing list.
- From the number of pallets, work out the number of transport packs to be sampled:
+ Check condition of boxes for integrity of packaging material.
+ Check for cleanliness of boxes.
+ Check the labelling of the boxes for damage.
+ Check the boxes for overall damage.
+ Check the labels for spelling mistakes.
+ Check the labels for manufacturing and expiry dates.
+ Count, categorize and record the number of defects.
- From the number of boxes selected, work out the number of unit packs to examined visually:
+ Check condition of the containers for integrity of packaging material.
+ Check for cleanliness of containers.
+ Check condition of containers for shape and colour.
+ Check the labelling of containers for damage.
+ Check the containers for overall damage.
+ Check the labels for spelling mistakes.
+ Check the labels for manufacturing and expiry dates.
+ Count, categorize and record the number of defects.
- From the number of containers selected, determine the number of containers to be taken for physical and chemical testing and for retention.
- Check the supplier certificate against the specifications, if applicable.
III. Types of sampling tools
Figure 1. Sampling scoops for solids

Figure 2. Typical dip tube
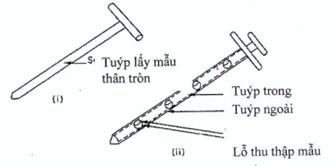
Figure 3. Typical sample thieves
(i) The plug thief (Figure 3.i) typically consists of a hollow tube with an inner rod that has a tip on the end to allow the thief to enter the powder bed in the closed position. Pointed tips distort the powder bed less than blunt-tipped probes. Some thieves have a locking device that allows the sample volume to be set to the required sample weight, thereby reducing the weight variation in the sample population.
(ii) A chamber thief (Figure 3.ii) generally consists of two concentric tubes; the inner tube is solid except for the chambers in which the sample is collected. The outer tube is hollow with openings that can be aligned with the chambers in the inner tube. A well-designed thief will have a sharp end to minimize disruption to the powder bed.
Note: When it is inserted into a static powder blend a thief will distort the bed by carrying pharmaceutical product from the upper layers of the blend to the lower layers. The magnitude of this distortion can depend on whether the thief is inserted into the blend with a smooth, jerky or twisting action. Therefore, staff should be trained in using the appropriate technique.
The angle at which the thief enters the powder bed can also influence sampling error. If a thief is inserted into the powder bed vertically, it can extract samples of different particle size from those that would be obtained using the same thief inserted at an acute angle. In addition, the orientation of a chamber thief in relation to the powder bed (i.e. whether the chamber is at the top, the bottom or in the middle of the thief) may also influence the sampling error.
Sampling error can also be affected by bed depth, as the static pressure of the bulk blend forces the material into the sample chamber(s). This pressure is far greater at the bottom of a large container than it is in the middle or at the top. It is quite possible that the same thief could extract samples of different particle size from the top or bottom of a static powder blend.
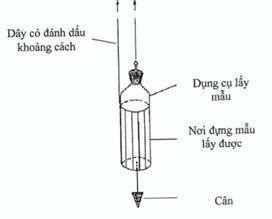
Figure 4. Weighted container
For taking samples from large tanks and storage vessels, a container in a weighted carrier can be used. The container is designed such that it can be opened at the required depth. Marks on the cord used for lowering the container can be used to determine when the correct
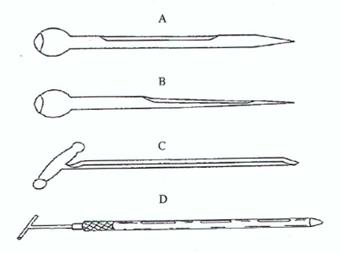
Figure 5. Typical sampling spears
A: Closed spear for sampling large grains such as maize
B: Closed spear for sampling small grains such as wheat
C: Open spear
D: Double-tube spear
Sampling spears generally have a maximum external diameter of about 12 mm, but can be up to 25 mm in diameter and should be 40–45 cm in length.
IV. Number of sampling units from batches of finished pharmaceutical products to be taken for visual inspection (ISO 2859-1)
| Lot size | Number of sampling units from batches of finished pharmaceutical products to be taken for visual inspection |
| 2 to 8 | 2 |
| 9 - 15 | 3 |
| 16 - 25 | 5 |
| 26 - 50 | 8 |
| 51 – 90 | 13 |
| 91 - 150 | 20 |
| 151 - 280 | 32 |
| 281 - 500 | 50 |
| 501 - 1200 | 80 |
| 1201 - 3200 | 125 |
| 3201 - 10000 | 200 |
| 10001 - 35000 | 315 |
| 35001 - 150000 | 500 |
| 150001 - 500000 | 600 |
| Over 500001 | 1250 |
V. Number samples taken for quality inspection
Number of samples of pharmaceutical products and pharmaceutical starting materials taken for quality inspection (excluding samples taken for retention purpose):
| No. | Dosage form | Type, specifications | Number |
| 1 | Tablets, capsules, film coated tablets | 1 active ingredient | 80 tablets/capsules |
| ≥ 2 active ingredients | 120 tablets/capsules | ||
| 2 | Solutions | ≥ 100 ml | 20 bottles (vials) |
| 10 - 100 ml | 30 bottles (vials) | ||
| 5ml - 10ml | 50 bottles (vials) | ||
| < 5ml | 100 bottles (vials) | ||
| 3 | Granules, powders | Packaged in single-dose or multiple-dose | ~ 100 gram |
| Hard pills, soft pills | > 0,5 g/pill | 120 pills | |
| 0,1 - 0,5 g/pill | 200 pills | ||
| < 0,1 g/pill | 500 pills | ||
| 4 | Medicinal liquor | ≤ 650 ml | 7 bottles |
| > 650 ml | 5 bottles | ||
| 5 | Intravenous fluids | ≥ 250 ml | 20 bottles |
| 100 ml - 250 ml | 25 bottles | ||
| < 100 ml | 50 bottles | ||
| Syringes | 1ml | 150 syringes | |
| ≥ 2 ml | 120 syringes | ||
| Distilled water for injection | 2 ml | 250 ampoules | |
| 5 ml | 100 ampoules | ||
| 10 ml | 80 ampoules | ||
| 6 | Eye drops | ≤ 2ml/100mg | 100 vials (tubes) |
| > 2ml/100mg | 80 vials (tubes) | ||
| 7 | Topical ointments, creams, gels | ≤ 100mg | 30 vials (tubes) |
| > 100mg | 40 vials (tubes) | ||
| 8 | Powders for injection | < 100 mg | 150 vials |
| 100 - 450 mg | 120 vials | ||
| > 450 mg | 100 vials | ||
| 9 | Massage oil | 1 - 2 ml | 30 vials |
| ≥ 5 ml | 20 vials | ||
| 10 | Medicinal extract | Various forms | ~100g |
| 11 | Herbal medicinal materials | Containing oil | 250 g |
| Not containing oil | 100 g | ||
| 12 | Oil | Various forms | 150 ml |
| 13 | Vaccines, biologicals | Various forms | In accordance with the manufacturer's regulations |
| 14 | Materials | Precious materials | 20 g |
| Antibiotic materials | 50 g | ||
| Materials for manufacture of narcotic drugs and psychotropic drugs, precursors | 10 g | ||
| Normal materials | 100 g | ||
| Plastic beads | 200 g | ||
| 15 | Infusion sets | Various forms | 30 sets |
| 16 | Hollow glass tubes | 2 ml | 500 tubes |
| ≥ 5 ml | 300 tubes | ||
| 17 | Bottles for intravenous fluids | Various forms | 10 bottles |
APPENDIX II
CLASSIFICATION OF VIOLATIONS AND CONCLUSIONS ON PHARMACEUTICAL PRODUCT RECALL
(Enclosed with the Circular No. 11/2018/TT-BYT dated May 04, 2018 of the Minister of Health)
I. First-degree violation means a violation where the pharmaceutical product threatens to cause serious harm to the users’ health or life in one of the following cases:
1. The pharmaceutical product is counterfeit, illegally imported and of unknown origins;
2. The pharmaceutical product contains substances banned from use in manufacture of pharmaceutical products;
3. The finished pharmaceutical product is manufactured from a material not intended for human use or material not yet granted licenses for use in manufacture of pharmaceutical products or food intended for human use;
4. The pharmaceutical product is manufactured at a facility not yet granted the certificate of eligibility for pharmacy business;
5. There is no evidence that the injection or parenteral pharmaceutical product has undergone quality inspection during the manufacture process and before release;
6. A foreign competent authority notifies a recall of the pharmaceutical product;
7. A competent authority concludes that the pharmaceutical product is not safe;
8. The pharmaceutical product contains wrong active ingredients;
9. The pharmaceutical product has wrong content that may lead to serious consequences;
10. The parenteral pharmaceutical product fails the sterility, pyrogen or endotoxin test;
11. The injection is not sterile;
12. The content, route of administration or dose on the label of the pharmaceutical product that contains a strong active ingredient and has low safety threshold is incorrect.
II. Second-degree violation means a violation where there is evidence that the pharmaceutical product does not guarantee effective treatment or is unsafe for users but does not cause harm to the users’ health or life in one of the following cases:
1. A competent authority concludes that the pharmaceutical product does not guarantee effective treatment;
2. The pharmaceutical product is manufactured from materials that fail to meet specifications;
3. There is no evidence that the pharmaceutical product has undergone quality inspection during the manufacture process and before release (except for the case specified in Clause 5 of Section II);
4. The pharmaceutical product is not granted the certificate of pharmaceutical product registration or the import license;
5. A competent authority concludes that the pharmaceutical product is granted the certificate of pharmaceutical product registration according to counterfeit documents;
6. The finished pharmaceutical product is manufactured from an expired pharmaceutical starting material or pharmaceutical starting material that has to be recalled as requested by a competent authority or pharmaceutical starting material of illegal origin (it is illegally imported or the material producer has not yet been granted the certificate of eligibility for pharmacy business);
7. The pharmaceutical product is manufactured at a manufacturing facility while it is being suspended or while the certificate of eligibility for pharmacy business is suspended;
8. The active ingredient content of the pharmaceutical product deviates by more than 5%;
9. The pharmaceutical product contains wrong active ingredients (except for the case in which the first-degree violation is committed);
10. The pharmaceutical product fails to meet specifications in terms of bacterial contamination (except for the cases specified in Clauses 10 and 11 Section II);
11. The injection or parenteral pharmaceutical product fails to meet specifications in terms of clarity, impurity, particles visible or invisible to the naked eye;
12. The tablet fails to meet specifications in terms of disintegration while it disintegrates in the acidic environment for more than 02 (two) hours (except for enteric-coated tablets);
13. The enteric-coated tablet containing active ingredients that are unstable or irritate stomach fails to meet specifications in terms of the disintegration or dissolution in the acidic environment;
14. The liquid injection has a volume smaller than 75% of that written on the label;
15. The powdered injection has a weight smaller than 75% of that written on the label;
16. The tablet has an average dissolution smaller than 50% than the permissible dissolution;
17. The pharmaceutical product fails to meet specifications in terms of relevant impurities;
18. The injection or parenteral pharmaceutical product fails to meet specifications in terms of pH;
19. The prolonged-release tablet and modified-release tablet fail to meet specifications in terms of dissolution;
20. The pharmaceutical product fails to meet specifications in terms of sedimentation of blend and emulsion for injection;
21. The pharmaceutical product is recalled by a foreign competent authority, except in the case it is urgently recalled, and is found to be imported into Vietnam through inspection;
22. The pharmaceutical product is incorrect due to mistakes during manufacture or labeling; the label of the pharmaceutical product specifies wrong route of administration, dosage, content, concentration of active ingredients and use (except for the case specified in Section I);
23. The pharmaceutical product is not manufactured and imported according to the application for pharmaceutical product registration or import license;
24. The pharmaceutical product contains content or concentration exceeding the permissible limits.
II. Third-degree violation means violations other than those specified in Sections I and II that do not affect the treatment ability and safety of the drug in one of the following cases:
1. The pharmaceutical product fails to meet specifications in terms of sensory analysis: color change; layer separation with respect to ointments, creams and gels;
2. The pharmaceutical product fails to meet specifications in terms of density;
3. The tablet fails to meet specifications in terms of weight variation tolerance (average weight of a tablet);
4. The ointment or cream fails to meet specifications in terms of weight variation tolerance;
5. The powdered injection has a weight greater than 75% of that written on the label but smaller than the registered limit;
6. The enteric-coated tablet fails to meet specifications in terms of disintegration but disintegrates for less than 02 (two) hours;
7. The sugar coated tablet and hard pill fail to disintegrate completely;
8. The tablet fails to meet specifications in terms of dissolution (except for the case specified in Clause 17 Section II);
9. The tablet fails to meet specifications in terms of content of active ingredients but the deviation of active ingredient content in the tablet is less than 5%;
10. The herbal tablet fails to meet specifications in terms of impurity and humidity;
11. The modern tablet, powdered injection and freeze-dried injection fail to meet specifications in terms of humidity;
12. The liquid pharmaceutical product fails to meet specifications in terms of pH (except for the case specified in Section II);
13. The oral solution fails to meet specifications in terms of sedimentation;
14. The oral solution and lotion fail to meet specifications in terms of volume;
15. The injection fails to meet specifications in terms of volume but its volume is not smaller than 75% of that written on the label;
16. The injection or parenteral pharmaceutical product fails to meet specifications in terms of pH;
17. The pharmaceutical product fails to satisfy all labeling requirements, except for the cases specified in Section I and II;
18. The pharmaceutical product packaging material and method fail to satisfy storage requirements;
19. The pharmaceutical product fails to satisfy average weight criterion, the pharmaceutical product is not manufactured according to the application for pharmaceutical product registration: change of weight of a tablet, excipient ratio and type of excipients.
IV. Other violations: The Drug Administration of Vietnam shall conclude the degree of violations after the certification advisory council of the Ministry of Health comments. Counsel of the council shall be given by assessing effects of the pharmaceutical product committing violations on users’ health.
APPENDIX III:
FORMS
(Enclosed with the Circular No. 11/2018/TT-BYT dated May 04, 2018 of the Minister of Health)
Form No. 01: Sample collection form
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
|
| (Place name), date (dd/mm/yyyy) …. |
FORM OF SAMPLE COLLECTION FOR QUALITY VERIFICATION
Letter of introduction or inspector card (specify number, date of issue and issuing authority): ……………………
Full names, position and workplace of people who took samples:
1 ................................................................................................................................
2 ................................................................................................................................
3 ................................................................................................................................
Name of the establishment where sample(s) was taken: ………………………………
Classification of the establishment where sample(s) was taken: ……………………………..
Address:…………………………….. Telephone: ……………………………..
| No. | Pharmaceutical product name, concentration, content, registration number | Batch number, date of manufacture, expiry date | Smallest pack | Quantity of sample taken | Name and address of manufacturer | Name of importer (in case of imported pharmaceutical product), distributor | Status of batch of pharmaceutical product prior to sampling |
|
|
|
|
|
|
|
|
|
Storage condition upon sampling: ………………….
This form is made into 03 copies: 01 copy is kept at the establishment where sample(s) was taken, 01 copy is kept at the testing authority, 01 copy is kept at ……….(authority in charge of quality inspection and management).
| Person(s) taking samples | Representative of the establishment where sample(s) was taken |
Form No. 02: Test report
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
TEST REPORT
No.
Sample to be tested:
Manufacturer:
Importer (in case of foreign pharmaceutical product):
Batch number: Date of manufacture: Expiry date:
Number of Certificate of registration or import license:
Place where sample was taken (sending sample):
Person taking sample (sending sample):
Testing requirements (specify contents, number and date of sample collection form or enclosed documents)
Date of receiving sample:
Test registration number:
Person delivering sample: Person receiving sample:
Standard applied:
Status of sample upon receipt and opening of seal for testing:
|
| Quality characteristics | Quality requirements | Results and conclusions |
| 1. |
|
|
|
| 2. |
|
|
|
| 3… |
|
|
|
Conclusion: (Specify whether the batch meets specifications; in case of failure to meet, specify reasons thereof).
|
| (Place name), date (dd/mm/yyyy) …. |
Form No. 03: Analysis report
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
ANALYSIS REPORT
No.
Sample to be analyzed:
Manufacturer:
Importer (in case of foreign pharmaceutical product):
Batch number: Date of manufacture: Expiry date:
Number of Certificate of registration or import license:
Place of sending sample:
Person sending sample:
Analysis requirements (specify contents, number and date of sample collection form or enclosed documents)
Date of receiving sample:
Analysis registration number:
Person delivering sample: Person receiving sample:
Standard applied:
Status of sample upon receipt and opening of seal for analysis:
| Quality characteristics | Quality requirements | Results |
| 1. |
|
|
| 2. |
|
|
| 3... |
|
|
|
| (Place name), date (dd/mm/yyyy) …. |
Form No. 04: Pharmaceutical product recall form
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
|
| (Place name), date (dd/mm/yyyy) …. |
PHARMACEUTICAL PRODUCT RECALL FORM
We are: (specify name and position of each member):
1/................................................................................................................................
2/................................................................................................................................
3/ ...............................................................................................................................
from ...........................................................................................................................
were assigned to recall pharmaceutical products that fail to meet specifications according to the Official Dispatch No. …………...... dated (date/month/year) .... of
We recalled the following pharmaceutical products at ……………………:
| No. | Pharmaceutical product name, concentration, content | Unit | Quantity of pharmaceutical products recalled | Batch number | Manufacturer | Notes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Representative of the establishment where pharmaceutical products were recalled | Members | Head of recall department |
Form No. 05: Pharmaceutical product recall report
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
|
| (Place name), date (dd/mm/yyyy) …. |
PHARMACEUTICAL PRODUCT RECALL REPORT
To:...
Implementing the Official Dispatch No. …………...... dated (date/month/year) .... of .... on recall of the pharmaceutical product..., registration number..., batch number..., date of manufacture..., expiry date... manufactured by... and imported by... (in case of imported pharmaceutical product),...(Name of the establishment) would like to submit a pharmaceutical product recall report. To be specific:
1. Information about the batch of recalled pharmaceutical product:
- Name of the pharmaceutical product, number of registration certificate or import license, name of active ingredient, concentration/content, dosage form, batch number, expiry date, manufacturer, importer;
- Time of release/import;
2. Recall results:
2.1. Result of recall of the pharmaceutical product from pharmacy business establishments:
| No. | Name of the trader purchasing pharmaceutical product | Unit | Quantity of pharmaceutical products purchased | Quantity of pharmaceutical products sold | Quantity of pharmaceutical products recalled | Notes |
| 1 |
|
|
|
|
|
|
| 2 |
|
|
|
|
|
|
| 3... |
|
|
|
|
|
|
| Total |
|
|
|
|
| |
2.2. Consolidation of recall results
- Quantity of pharmaceutical products manufactured/imported;
- Quantity of pharmaceutical products sold on the market;
- Quantity of pharmaceutical products recalled.
|
| (Place name), date (dd/mm/yyyy) …. |
Form No. 06: Pharmaceutical product destruction form
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
| No. | (Place name), date (dd/mm/yyyy) …. |
PHARMACEUTICAL PRODUCT DESTRUCTION FORM
Implementing the Decision No. …….. dated (date/month/year) … of ……… on destruction of pharmaceutical products that fail to meet specifications and expire.
Today, on (date/month/year) … at (name of the place where pharmaceutical products are destroyed): …………
The pharmaceutical product destruction council established according to the Decision No. …….. dated (date/month/year) … of ……… is composed of:
1 ................................................................................................................................
2 ................................................................................................................................
3 ................................................................................................................................
...................................................................................................................................
and have witnessed and carry out destruction of the following pharmaceutical products:
| No. | Pharmaceutical product name, concentration, content | Batch number | Name of manufacturer | Planned quantity of pharmaceutical products destroyed | Actual quantity of pharmaceutical products destroyed | Difference | Notes |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
((*) If there is difference between the actual and planned quantity of pharmaceutical products destroyed, explanation thereof must be provided)
Destruction method:
...................................................................................................................................
...................................................................................................................................
The pharmaceutical product destruction form is sent to .................................................
This form is made into ….. copies, each of which is kept by a party and ……. copies are sent.
| Members destroying pharmaceutical products | President of the pharmaceutical product destruction council |
Form No. 07: Report on sampling of pharmaceutical products for quality inspection
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
| No. | (Place name), date (dd/mm/yyyy) …. |
REPORT ON SAMPLING OF PHARMACEUTICAL PRODUCTS FOR QUALITY INSPECTION
To: The Drug Administration of Vietnam - The Ministry of Health
| Name and address of manufacturer | Name of manufacturing country | Name of pharmaceutical product (dosage form), active ingredients, content | Registration number or import license number | Batch number, date of manufacture (if any), expiry date | Packaging specification/ Unit (smallest pack) | Quantity of pharmaceutical products imported (*) | Name and telephone of the applicant | Name and telephone of the importer | Name and telephone of the establishment entrusted to import | Name and telephone of Class I distributor (if any) | Date of import (*) | Name and telephone of sampling facility and testing facility | Date of sampling | Date of issuing test report | Test result (satisfactory/ unsatisfactory) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(*) The sampling facility and testing facility are not required to report these contents.
|
| ...,(Place name), date (dd/mm/yyyy) …. |
Form No. 08: Certificate of vaccine/biological quality
| Name of the supervisory authority | THE SOCIALIST REPUBLIC OF VIETNAM |
CERTIFICATE OF VACCINE/BIOLOGICAL QUALITY
No.
| Commercial name: Common name: | Number of Certificate of registration or import license: Quantity sample of the testing facility: |
| Batch No. (on the vial/syringe/phial): | Batch No. (on the box): |
| Date of manufacture: | Expiry date: |
| Packaging method: | Sterile diluent batch No. (if any): Expiry date: |
| Manufacturer: | Importer: |
| Date of manufacture/ import: | Quantity vaccines/biologicals manufactured/imported: |
Conclusion:
(Specify whether the batch satisfies specifications approved by the Ministry of Health or whether the batch satisfies requirements for document inspection and storage conditions in the cold chain during the import. In case of failure to satisfy, specify reasons thereof).
|
| (Place name), date (dd/mm/yyyy) …. |
- 1 Circular No. 09/2010/TT-BYT of April 28, 2010, guiding the management of medicine quality
- 2 Circular No. 03/2020/TT-BYT dated January 22, 2020 on amendments to Circular No. 11/2018/TT-BYT on quality of pharmaceutical products and pharmaceutical starting materials
- 3 Integrated document No. 06/VBHN-BYT dated July 03, 2020 Circular on quality of pharmaceutical products and pharmaceutical starting materials
- 4 Integrated document No. 06/VBHN-BYT dated July 03, 2020 Circular on quality of pharmaceutical products and pharmaceutical starting materials
- 1 Circular No. 06/2018/TT-BYT dated April 06, 2018 promulating the nomenclature of imported and exported drugs and medicinal ingredients for human use and cosmetics whose HS codes have been assigned in the Vietnam’s nomenclature of exports and imports
- 2 Circular No. 03/2018/TT-BYT dated February 09, 2018 Good Distribution Practices for pharmaceutical products and pharmaceutical starting materials
- 3 Circular No. 01/2018/TT-BYT dated January 18, 2018
- 4 Decree No. 75/2017/ND-CP dated June 20, 2017, defining functions, tasks, powers and organizational structure of Ministry of Health
- 5 Decree No. 54/2017/ND-CP dated May 08, 2017, guidelines for implementation of the Law on Pharmacy
- 6 Law No. 105/2016/QH13 dated April 06th, 2016, on pharmacy
- 7 Law No. 01/2011/QH13 of November 11, 2011 on Archives
- 1 Circular No. 06/2018/TT-BYT dated April 06, 2018 promulating the nomenclature of imported and exported drugs and medicinal ingredients for human use and cosmetics whose HS codes have been assigned in the Vietnam’s nomenclature of exports and imports
- 2 Circular No. 03/2018/TT-BYT dated February 09, 2018 Good Distribution Practices for pharmaceutical products and pharmaceutical starting materials
- 3 Circular No. 01/2018/TT-BYT dated January 18, 2018