
PHỤ GIA THỰC PHẨM - PHƯƠNG PHÁP XÁC ĐỊNH CÁC THÀNH PHẦN VÔ CƠ
Food additives - Methods for determining inorganic components
Lời nói đầu
TCVN 6468 : 1998 hoàn toàn phù hợp với phần II của sách Hướng dẫn yêu cầu kỹ thuật cho những chú ý chung, thử nhận biết, dung dịch thử và các tài liệu tham khảo khác của JECFA (Guide to specifications, general notices, general analytical techniques, identification tests, test solutions and other reference materials – JECFA - FAO FOOD and nutrition paper - 5 rev 2).
TCVN 6468 : 1998 do ban kỹ thuật tiêu chuẩn TCVN/TC/F4 Phụ gia thực phẩm biên soạn, Tổng cục Tiêu chuẩn - Đo lường - Chất lượng đề nghị, Bộ Khoa học, Công nghệ và Môi trường ban hành.
PHỤ GIA THỰC PHẨM - PHƯƠNG PHÁP XÁC ĐỊNH CÁC THÀNH PHẦN VÔ CƠ
Food additives - Methods for determining inorganic components
Tiêu chuẩn này quy định phương pháp để xác định các thành phân vô cơ trong phụ gia thực phẩm.
Chuyển 2 g mẫu, đã được cân chính xác, vào một cốc 250 ml chứa 150 ml nước và 1,5 ml axit sunfuric đậm đặc. Đậy cốc bằng một nắp kính đồng hồ và đun nóng hỗn hợp trên một bình cách thuỷ trong 6 giờ, thường xuyên cọ thành cốc bằng một que khuấy đầu bịt cao su và thêm nước đã bị hao do bốc hơi. Cân 500 mg chính xác tới 0,1 mg một chất trợ lọc thích hợp đã được sấy khô từ trước ở 105°C trong 1 giờ, cho vào dung dịch mẫu và lọc qua một chén nung Gooch (đã cân trước), có một tấm đệm amiăng. Rửa cặn còn lại một vài lần bằng nước nóng, sấy khô chén và cặn ở 105°C trong 3 giờ, để nguội trong bình hút ẩm và cân. Chênh lệch giữa khối lượng tổng cộng với khối lượng của chất trợ lọc cộng với chén nung và tấm đệm là khối lượng của chất không tan trong axit. Tính phần trăm theo khối lượng.
2.1 Xác định tổng số1
Cân chính xác khoảng 3 g mẫu trừ khi có chỉ dẫn khác trong một chén nung đã biết khối lượng, đốt ở nhiệt độ thấp (khoảng 550°), không vượt quá màu đỏ thật xỉn, cho đến kho hết than, để nguội trong bình hút ẩm, và cân. Nếu không thu được tro trắng thì làm ướt khối mẫu đã than hoá này bằng nước nóng, thu lấy cặn không tan lên một giấy lọc không tro và đốt phần cặn này và giấy lọc, cho đến khi tro có mầu trắng hoặc gần như trắng. Cuối cùng, thêm dịch lọc, sau đó làm bốc hơi tới khô, và đem nung toàn bộ tới màu đỏ xỉn. Nếu vẫn không thu được tro trắng, thì làm nguội chén nung, cho thêm 15 ml etanol, dùng que thuỷ tinh làm vụn tro, sau đó đốt hết etanol, lại nung toàn bộ tới màu đỏ xỉn, để nguội trong bình hút ẩm và cân.
Chú thích - Nếu có khó khăn với việc oxy hoá các chất hữu cơ, việc sử dụng một chất trợ tro như ammonium nitrat có thể chứng tỏ là tốt hơn so với việc hoà tan phần cặn và đem lọc trước khi tro hoá tiếp theo.
2.2 Xác định tro không tan trong axit
Đun sôi tro đã thu được như chỉ dẫn đối vơi tro tổng số ở trên, với 25 ml axit clohydric loãng ST trong 5 phút, thu lấy chất không tan trên một giấy lọc không tro thích hợp, rửa bằng nước nóng, đốt ở 800°±25°, để nguội và cân. Tính phần trăm của tro không tan trong axit theo khối lượng của mẫu thử.
2.3 Xác định tro (tro sunfat hoá)
2.3.1 Phương pháp I (đối với chất rắn)
Chuyển một lượng mẫu thử đã chỉ dẫn trong các chuyên luận riêng vào một đĩa platin 50 ml - 100 ml đã biết khối lượng hay một dụng cụ thích hợp khác và thêm đủ axit sunfuric loãng ST để thấm ướt toàn bộ mẫu. Nung nóng từ từ, bằng cách dùng một bết điện, một bếp Argand hay đèn đốt hồng ngoại, cho đến khi mẫu khô và hoàn toàn hoá than, sau đó tiếp tục gia nhiệt cho đến kho toàn bộ mẫu bay hơi hoặc gần như tất cả cacbon đã bị oxy hoá, và để nguội, tiếp đó thấm ướt cặn này bằng 0,5 ml axit sunfuric đậm đặc và đem đốt theo cùng cách như trên cho đến khi phần dư của mẫu và mọi axit thừa bay hơi hết. Cuối cùng, nung trong lò nung ở nhiệt độ 800° ± 25° trong 15 phút hay lâu hơn, nếu cần, để hoàn tất việc nung cuối cùng rồi để nguội trong bình hút ẩm, và cân.
Chú thích - Để thúc đẩy quá trình bốc hơi axit sunfuric, nên cho thêm một vào mẩu ammoni cacbonat ngay trước khi hoàn thành việc nung.
2.3.2 Phương pháp II (đối với chất lỏng)
Trừ khi có các chỉ dẫn khác, nếu không thì chuyển một lượng mẫu theo yêu cầu vào một cốc chứa thích hợp đã biết khối lượng, thêm 10 ml axit sunfuric loãng ST, và trộn kỹ. Làm bốc hơi mẫu triệt để bằng cách đun nhẹ, không làm sôi, và để nguội. Cuối cùng, nung nóng lò nung ở nhiệt độ 800° ± 25° trong 15 phút hoặc lâu hơn, để nguội trong bình hút ẩm, và cân.
Trừ khi có các chỉ dẫn khác, nếu không thì cho một lượng mẫu thử đã quy định vào một ống nghiệm Nessler, hoà tan trong khoảng 30 ml nước, và trung hoà bằng axit nitric loãng ST nếu dung dịch là kiềm. Thêm 6 ml axit nitric loãng và pha loãng ST bằng nước với 50 ml. Nếu sử dụng dung dịch mẫu thử đã xử lý, thì chuyển dung dịch mẫu vào ống nghiệm Nessler và pha loãng bằng nước tới 50 ml. Chuyển một lượng đã định axit clohydric 0,01 N vào một ống nghiệm Nessler khác để làm chuẩn, thêm 6 ml axit nitric loãng ST và pha loãng bằng nước tới 50 ml.
Nếu dung dịch chứa mẫu không trong, lọc cả hai dung dịch trong cùng một điều kiện. Cho thêm 1 ml thuốc thử nitrat bạc ST vào từng dung dịch, trộn kỹ, và để yên trong 5 phút, tránh ánh sáng mặt trời trực tiếp. So sánh độ đục của hai dung dịch bằng cách quan sát các ống Nessler từ thành ống và từ trên xuóng tương phản với một nền mầu đen. Độ đục của dung dịch mẫu không được vượt quá độ đục của dung dịch chuẩn.
4.1 Phương pháp I
Phương pháp đo mầu thori nitrat
Nên dùng phương pháp này, trừ khi có những chỉ dẫn khác trong chuyên luận riêng.
Cảnh báo : Khi áp dụng phép thử này đối với các hợp chất hữu cơ, nhiệt độ ở nơi tiến hành chưng cất phải được kiểm soát chặt chẽ trong suốt thời gian thí nghiệm ở dải yêu cầu từ 135° đến 140° để tránh khả năng nổ.
Chú thích - Để giảm tối thiểu việc chưng cất mẫ trắng bị rò rỉ flo từ dụng cụ thuỷ tinh, dụng cụ chưng cất cần được xử lý như sau : Xử lý đồ thuỷ tinh bằng dung dịch natri hydroxit 10 %, sau đó dùng nước ở vòi xối rửa và tráng bằng nước cất. Ngoài ra, ít nhất ngày một lần, xử lý bằng cách đun sôi 15 - 20 ml axit sunfuric loãng (1 : 2) cho đến khi thiết bị chưng cất chứa đầy khói; để nguội, đổ axit ra, xử lý lại với dung dịch natri hydroxit 10 % và rửa tráng kỹ. Về chi tiết xem mục 25.050 và 25.054 trong các phương pháp phân tích chính thức của AOAC, xuất bản lần thứ 13, năm 1980.
Trừ khi có các chỉ dẫn khác, nếu không thì cho 0,5 g mẫu và 30 ml nước vào một vình chưng cất dung tích 125 ml, có một ống bên và ống nhận. Bình chưng cất được nối với một bộ phận ngưng tụ và có một nhiệt kế và một ống mao dẫn, cả hai bộ phận này phải chạm vào chất lỏng. Cho từ từ 10 ml axit pecloric trong khi khuấy liên tục, sau đó cho thêm 2 hay 3 giọt dung dịch nitrat bạc (1 : 2) và một vài hạt bi thuỷ tinh, Nối một phễu nhỏ giọt nhỏ hay một nguồn tạo hơi với ống mao dẫn. Đặt bình chưng cất trên một tấm amiăng có lỗ đảm bảo cho khoảng 1/3 bình chưng cất tiếp xúc với ngọn lửa. Chưng cất cho đến khi nhiệt độ đạt tới 135°.
Cho thêm nước từ phễu hay xông hơi qua ống mao dẫn, duy trì nhiệt độ trong khoảng 135° đến 140° trong suốt thời gian thí nghiệm. Tiếp tục chưng cất cho đến khi thu được 100 ml dịch cất. Sau khi thu được 100 ml (dịch cất A), lấy thêm một lượng 50 ml (dịch cất B) để đảm bảo rằng tất cả flo đã được bốc hơi hết.
Cho 50 ml dịch cất A vào một ống Nessler dung tích 50 ml. Trong một ống Nessler tương tự khác cho 50 ml nước cất từ thiết bị này để đối chứng. Cho thêm vào mỗi ống 0,1 ml dung dịch natri alizarinsulfonat (1 : 1.000) đã lọc và 1 ml dung dịch hydroxylamin hydroclorua mới được chuẩn bị (1 : 4.000) và trộn kỹ. Thêm từng giọt và khuấy dung dịch hydroxid natri 1 N hoặc 0,05 N, số lượng tuỳ thuộc vào thể tích flo lượng dự kiến bốc hơi và chưng cất dư thừa vào ống chứa thành phẩm chưng cất cho đến khi mầu của nó vừa trùng với mầu kiểm soát, đó là mầu hồng nhạt. Sau đó thêm vào từng ống 1,0 ml axit clohydric 0,1 N và trộn kỹ. Dùng một buret có khắc độ tới 0,05 ml, thêm từ từ vào ống nghiệm chứa dich cất đủ một lượng dung dịch thori nitrat (1 : 4000) sao cho sau khi trộn lẫn, mầu của chất lỏng chuyển ngay thành mầu hồng nhạt. Ghi lại lượng dung dịch đã thêm vào, sau đó thêm chính xác cùng một lượng như vậy vào mẫu kiểm tra (đối chứng), và trộn. Bây giờ cho thêm vào mẫu kiểm tra dung dịch natri florua (10 μg F/ml) bằng một buret để tạo mầu của hai ống này trung hợp với nhau sau khi pha loãng tới cùng một thể tích. Trộn kỹ, và để cho tất cả các bọt khí thoát ra trước khi tiến hành so sánh mầu lần cuối. Kiểm tra điểm kết thúc bằng cách cho thêm 1 hoặc 2 giọt thuốc thử natri florua vào mẫu kiểm tra. Sự khác biệt rõ ràng về mầu sắc sẽ xẩy ra. Ghi lại khối lượng thuốc thử natri florua đã thêm.
Pha loãng dịch cất B thành 100 ml, và trộn kỹ. Cho 50 ml của dung dịch này vào một ống Nessler 50 ml, và theo các bước tiến hành như đối với dịch A. Lượng thuốc thử natri florua tổng cộng cần cho các dung dịch từ cả hai dịch cất A và B không nên vượt quá 2,5 ml.
4.2 Phương pháp II
Phương pháp A điện cực chọn lọc ion
4.2.1 Dung dịch đệm
Pha loãng 36 g axit cyclohexylenendinitrilo - tetra - axetic (CDTA) trong một lượng đủ hydroxit 1 M đến vừa đủ 200 ml. Chuyển 20 ml dung dịch này (tương đương với 4 g dinatri CDTA) vào một cốc 1.000 ml có chứa 500 ml nước, 57 axit axetic băng, và 58 g natri clorua và khuấy để hoà tan. Điều chỉnh pH của dung dịch tới khoảng 5,0 đến 5,5 bằng cách cho thêm dung dịch hydroxit - natri 5 M, sau đó làm nguội tới nhiệt độ phòng, pha loãng đến 1.000 ml bằng nước rồi trộn.
4.2.2 Cách tiến hành
Trừ khí có các chỉ dẫn khác trong từng chuyên luận, nếu không thì cho 8,0 g mẫu và 20 ml nước vào một bình chưng cất 250 ml, cho thêm 20 ml axit pecloric một cách thận trọng và sau đó thêm 2 đến 3 giọt dung dịch nitrat bạc (1 : 2) và một vài hạt bi thuỷ tinh. Theo các chỉ dẫn và xem kỹ các lưu ý và cảnh báo đã nêu trong phương pháp I, chưng cất dung dịch này cho đến khi thu được 200 ml dịch cất.
Chuyển 25,0 ml dịch cất vào một cốc nhựa 250 ml và pha loãng tới 100 ml bằng cách dung dịch đệm. Đưa các điện cực so sánh và ion flo (hoặc một điện cực flo hỗn hợp) của một thiết bị cực ion chọn lọc (như là thiết bị Orion model 407) vào dung dịch, và chỉnh bộ phận hiểu chỉnh cho đến khi kim báo chỉ vào điểm trung tâm của thang nồng độ Lô-ga-rit, để đủ thời gian cho cân bằng (khoảng 20 phút) và khuấy đều trong thời gian cân bằng và trong suôts thời gian còn lại của thí nghiệm. Dùng pipet nhỏ 1,0 ml dung dịch có chứa 100 μg ion flo (F) trong 1 ml (được chuẩn bị bằng cách hoà tan 22,2 mg natri florua, đã được sấy khô từ trước ở 200°C trong 4h, trong một lượng nước đủ để tạo 100,0 ml) vào trong một cốc, để cho điện cực đạt tới mức cân bằng và ghi lại số đọc cuối cùng trên thang nồng độ Lô-ga-rít (chú thích phải theo đúng các chỉ dẫn của nhà chế tạo thiết bị, lưu ý đến các cảnh báo và những yếu tố gây nhiễu, nạp điện lực và kiểm tra nhiệt độ, bộ phận bù nhiệt, và hiệu chỉnh).
4.2.3 Tính toán
Tính hàm lượng flo, theo miligam trên kilogam, theo công thức :
[IA/ (R - I)] x 100 x [200 / 25W]
Trong đó
I là số đọc trên thang ban đầu, trước khi thêm dung dịch natri florua;
A là nồng độ, flo trong dung dịch natri florua đã thêm vào dung dịch mẫu;
R là số đọc trên thang cuối cùng, sau khi đã thêm dung dịch natri florua, tính bằng μg trên ml;
W là lượng ban đầu của mẫu, tính bằng gam.
4.3 Phương pháp III
Phương pháp B điện cực chọn lọc Ion
4.3.1 Dung dịch natri florua (5 μg F trong ml) :
Chuyển 2,210 g natri florua, đã được sấy khô từ trước ở nhiệt độ 110° trong 2 giờ và đã được cân chính xác, cho vào một cốc nhựa 400 ml, thêm 200 ml nước, khuấy cho đến khi hoà tan. Cho toàn bộ dung dịch này vào một bình định mức 1000 ml, thêm nước pha loãng tới vạch mức và trộn đều. Bảo quản dung dịch gốc này trong một lọ nhựa. Vào ngày thí nghiệm, chuyển 5,0 ml dung dịch gốc này vào một bình định mức 1000ml, pha loãng bằng nước tới vạch mức và trộn đều.
4.3.2 Đường cong chuẩn
Chuyển vào từng cốc nhựa dung tích 250 ml các lượng 1,0; 2,0; 3,0; 5,0; 10,0 và 15,0 ml, dung dịch natri florua thêm 50 ml nước, 5 ml axit clohydric 1 N, 10 ml natri xitrat 1 M, và 10 ml natri EDTA 0,2 M vào mỗi cốc và trộn kỹ. Chuyển từng dung dịch vào một bình định mức 100 ml, pha loãng bằng nước tới vạch và trộn kỹ. Chuyển 50 ml của mỗi dung dịch còn lại vào một cốc nhựa dung tích 125 ml và đo điện thế của từng dung dịch với một thiết bị điện cực ion chọn lọc thích hợp (như thiết bị Orion Model No. 94-09, với màng thể rắn), dùng một điện cực so sánh thích hợp (như Orion Model No. 90-01 với đầu nối đơn). Vẽ đường cong hiệu chuẩn trên giấy bán Lô-ga-rít hai chiều (như loại giấy K & E No. 465130) với dung dịch μg F trên 100 ml dung dịch trên thang tỷ lệ Lô-ga-rít.
4.3.3 Cách tiến hành
Chuyển 1,00 g mẫu vào một cốc thuỷ tinh dung tích 150 ml, khuấy đều trong khi cho thêm 10 ml nước cho thêm từ từ 20 ml axit clohydric 1 N để hoà tan mẫu. Đun sôi nhanh trong 1 phút, sau đó chuyển vào một cốc nhựa 250 ml, và làm nguội nhanh trong nước đá. Thêm 15 ml natri xitrate 1 M và 10 ml dinatri EDTA 0,2 M rồi trộn kỹ. Chỉnh pH tới 5,5 ± 0,1 bằng axit clohydrric 1 N hay hydroxit natri 1 M, nếu cần, sau đó chuyển vào một bình định mức 100 ml, pha loãng tới vạch bằng nước và trộn kỹ. Chuyển 50 ml dung dịch này còn lại vào một cốc nhựa 125 ml và đo điện thế của dung dịch này bằng thiết bị đã mô tả ở trên theo đường cong chuẩn. Xác định hàm lượng florua, tình bằng μg, của mẫu thử từ đường cong chuẩn.
4.4 Phương pháp IV
Phương pháp C điện cực chọn lọc ion
4.4.1 Dung dịch đệm A
Thêm 2 thể tích axit axetic 6 N vào 1 thể tích nước, và chỉnh pH tới 5,0 bằng dung dịch hydroxit kali 50 %.
4.4.2 Dung dịch đệm B
Hoà tan 150 g natri xitrat dihydrat và 10,3 g dinatri EDTA dihydrat vào 800 ml nước, chính pH tới 8,0 bằng dung dịch hydroxit natri 50 %, và dùng nước pha loãng tới 1000 ml.
4.4.3 Dung dịch đệm C
Hoà tan bằng cách đun sôi 36 g axit cyclohexylendinitrilo - tetra - axetic (CDTA) trong một lượng hydroxit natri 1 N đủ để được 200 ml, sau đó để nguội, và lọc qua giấy lọc sợi thuỷ tinh. Dùng pipet cho 30 ml dung dịch này vào một hỗn hợp bao gồm 750 ml nước, 87 g natri clorua, và 85,5 ml axit axetic băng. Chỉnh pH tới khoảng 5,0 đến 5,5 bằng cách thêm dung dịch natri hydroxit 50 %, sau đó để nguội, và dùng nước pha loãng tới 3,000 ml.
4.4.4 Dung dịch chuẩn flo
Dùng một dung dịch có chứa 100 μg ion flo (F) trong 1 ml (100 mg/kg) có bán trên thị trường, hoặc được chuẩn bị bằng cách hoà tan 22,2 mg natri florua, đã sấy khô trước ở nhiệt độ 200° trong 4 giờ, vào một lượng nước đến vừa đủ 100,0 ml.
4.4.5 Chuẩn bị mẫu
Cân chính xác một lượng mẫu đã xác định trong chuyên luận, chuyển vào một bình đinh mức 100 ml, và hoà tan với lượng nước ít nhất, hoặc trong một thể tích dung dịch axit clohydric, đã được xác định trong chuyên luận. Thêm 50,0 ml dung dịch đệm thích hợp A, B hay C như đã xác định trong chuyên luận này, thêm nước (pha loãng) tới vạch mức và trộn kỹ.
4.4.6 Cách tiến hành
Dùng pipet hút 50 ml mẫu đã chuẩn bị vào một cốc nhựa và cho các điện cực so sánh và ion flo (hoặc moọt điện cực flo kết hợp) của một thiết bị điện cực chọn lọc ion phù hợp, với máy khuấy từ (Orion Model 407 hay tương đương) vào dung dịch. Bắt đầu cho khuấy từ thường, và đặt độ lệch của thước đo tới 100 % và núm kiểm soát nhiệt độ tới nhiệt độ phòng, nhiệt độ này phải bằng nhiệt độ dung dịch. Điều chỉnh bộ phận hiểu chuẩn để có thể đọc bất cứ số đo nào trên thang Lô-ga-rít tăng dần, và để cho thiết bị đo ổn định.
Chú thích - Điện cực chọn lọc ion phản ứng chậm hơn nhiều so với điện cực pH, và có thể không nhận được một số đo ổn định trong vòng 2 - 3 phút. Số đo này cần phải không được thay đổi trong 30 – 60 giây.
Thêm chính xác một lượng dung dịch chuẩn flo, như đã ghi trong các chuyên luận, để điện cực đạt tới trạng thái cân bằng trong khi vẫn tiếp tục khuấy, và lấy số đọc cuối cùng trên thang Lô-ga-rít tăng dần, ghi lại giá trị nhận được là S. Thực hiện phép thử trắng bằng cách dùng 50 ml dung dịch đệm cùng loại như đã dùng đối với mẫu đang phân tích và ghi giá trị nhận được này là B.
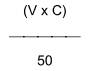 4.4.7 Tính toán
4.4.7 Tính toán
(V x C) Xác định giá trị ∆ theo công thức
Trong đó :
V là thể tích dung dịch flo chuẩn được thêm vào, tính bằng ml;
C là nồng độ chính xác của dung dịch flo chuẩn, tính bằng mg/kg;
50 là lượng (ml) mẫu đã chuẩn bị được sử dụng.
Tính toán Nồng độ flo (F) trong mẫu bằng miligam trên kilogam, theo công thức :
[(S x ∆) - B] x (100 / W)
trong đó :W là lượng mẫu đã lấy, tình bằng g.
5 Xác định lượng hao hụt sau khi sấy
Chú thích - Cần thực hiện các bước phòng ngừa phụ thích hợp khi cân các mẫu dễ hút ẩm hay tan rữa để đảm bảo ràng chúng không hút thêm ẩm.
Trừ khi có các chỉ dẫn khác trong chuyên luận riêng, nếu không, tiến hành việc xác định với 1 g - 2 g chất thử, đã được trộn kỹ từ trước và cân chính xác. Làm nhỏ mẫu tới mức bột mịn khi mẫu là các hạt tinh thể. Cân một chén cân nông có nắp thuỷ tinh, đã được sấy khô trong 30 phút trong cùng một điều kiện như sấy mẫu thử. Cho mẫu vào trong chén, đậy nắp, và cân. Dàn đều mẫu thử tới độ dầy khoảng 5 mm, và không quá 10 mm trong trường hợp là các vật liệu lớn. Đặt chén có mẫu vào buồng sấy, mở nắp và cũng để nắp trong buồng sấy, và sấy mẫu ở nhiệt độ và trong thời gian đã quy định theo tài liệu riêng, khi mở lò sấy, ngay lập tức đóng nắp chén cân lại và đặt nó nguội tới nhiệt độ phòng, trong một bình hút ẩm trước khi cân.
Nếu chất thử nóng chảy ở một nhiệt độ thấp hơn nhiệt độ đã xác định cho việc thử này, chuẩn bị mẫu như đã mô tả ở trên, sau đó đặt nó vào một bình hút chân không có chứa axit sunfuric. Rút chân không tới áp suất 130 Pa (1 mm thuỷ ngân), duy trì chân không này trong 24 giờ, và sau đó cân mẫu khô.
6 Xác định lượng hao hụt sau khi nung
Tiến hành như chỉ dẫn trong mục “lượng hao hụt sau khi sấy“. Tuy nhiên, trừ khi có các chỉ dẫn khác, nếu không, đốt mẫu ở nhiệt độ 450° tới 550° và dùng đĩa platin, thạch anh hoặc sứ thay cho chén cân.
7 Xác định các tạp chất kim loại
Tất cả các phân tích xác định các vết kim loại, đều bắt đầu bằng việc hoà tan mẫu và, nếu có thể được, bằng việc phân huỷ các chất hữu cơ trong mẫu thử. Hàm lượng vết kim loại khi đó có thể được xác định bằng các phương pháp đo trên thiết bị hay bằng phương pháp hoá học.
Phương pháp phổ nguyên tử kết hợp có thể xác định nhanh và độ chính xác cao, hiện đang được sử dụng rộng rãi trong trường hợp có một số lượng lớn mẫu cần thử.
Phương pháp hoá học dựa trên sự hình thành các hợp chất mầu đặc trưng của các tạp chất kim loại. Cường độ mầu của mẫu và của các chuẩn khi đó được so sánh bằng mắt hoặc dùng một quang phổ kế.
8 Các phương pháp sử dụng thiết bị (dự kiến)
Các phương pháp được môt ta trong mục này được coi là các ví dụ về phương pháp đo trên thiết bị đo, có thể dùng để phân tích đinh lượng một vài tạp chất kim loại trong các phụ gia thực phẩm. Cũng có thể sử dụng các phương pháp khác đã được thừa nhận.
Phương pháp I có thể áp dụng đối với các chất tan được trong axit loãng hoặc hỗn hợp axit. Phương pháp II được dùng cho các chất khác. Việc lựa chọn phương pháp cho xử lý ban đầu của một chất có thể theo các chỉ dẫn đã cho trong các chuyên luận riêng về phép thử giới hạn kim loại nặng.
8.1 Nguyên tắc
Mẫu thử được hoà tan trong axit hoặc được vô cơ hoá trong một hỗn hợp axit sunfuric, axit nitric và trong một số trường hợp là axit pecloric. Các nguyên tố bari, cadimi, chì, đồng, crôm, và kẽm trong dung dịch được xác định bằng phương pháp phổ hấp thụ nguyên tử ngọn lửa thông thường. Antimoan và aseni được xác định bằng cách sử dụng kỹ thuật tạo hydrua lại nhạy hơn.
8.2 Lưu ý chung
Do lượng kim loại nhỏ, nên đòi hỏi hết sức cẩn thận để giảm mức thuốc thử tới mức thấp nhất để tránh gây ô nhiễm trong khi thử. Mọi dụng cụ thử phải được làm sạch hoàn toàn bằng hỗn hợp axit loãng nóng (1 phần axit clohydric, 1 phần axit nitric đặc và 3 phần nước), sau đó rửa sạch ngay bằng nước trước khi sử dụng.
8.3 Thiết bị dụng cụ
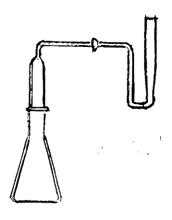
8.3.1 Bình kendal, làm bằng thuỷ tinh silic hay thuỷ tinh bosilicat (dung tích danh định 100 ml) được lắp thêm ở cổ một khớp nối B24 như hình 1. Phần nối thêm này được dùng để ngưng khói và có mang một phễu có khoá qua đó có thể đưa các thuốc thử vào bình.
Hình 1 - Bình kendal cải tiến (kiểu hở)
8.3.2 Thiết bị quang phổ hấp thụ nguyên tử. Có thể dùng bất kỳ thiết bị nào có bán trên thị trường vận hành theo kiểu hấp thụ, với điều kiện nó có các phương tiện để lựa chọn hỗn hợp chất oxy hoá / nhiên liệu cần thiết từ việc lựa chọn không khí, khi agonm oxit nitơ, hydro và axetylen, và có độ dài bước sóng từ 180 đến 600 nm.
Bộ phụ tùng tạo hydrua cũng cần thiết và hiện có sẵn ở các hãng chế tạo thiết bị hấp thụ nguyên tử. Để thao tác theo kiểu phát xạ và các phép đo hấp thụ có liên quan đến việc tạo ra một hydrua thể khí, khi cần có một bộ ghi điện thế, tốt hơn cả nên dùng loại nhiều dải, phủ được dải từ 1 - 20 mV.
8.4.3 Thuốc thử
Thuốc thử phải là loại tinh khiết cao hơn cấp chất lượng thuốc thử phân tích. Hoàn toàn dùng nước không chứa kim loại (xem ở dưới) :
(a) Axit nitric, tỷ trọng 1,42
(b) Axit peclỏic, dung dịch 60 % (w/w)
(c) Axit sunfuric, H2SO4 98 %
(d) Axit clohydric, tỷ trọng 1,16 - 1,18
(e) Dung dịch axit clohydric 5 N được chuẩn bị băng cách pha loãng thuốc thử (d) với nước cất không chứa kim loại
(f) Nước không chứa kim loại. Nước cất có thể được chưng cất lại từ một thiết bị làm bằng thuỷ tinh hoàn toàn, hoặc có thể cho đi qua một cột nhựa trao đổi cation, ví dụ như Amberlite IR 120 (H)
(g) Natri sunfat
(h) Các hạt nhỏ natri borohydrua
(i) Kali clorua
8.5 Dung dịch chuẩn :
Sử dụng dung dịch chuẩn bán sẵn trên thị trường, hoặc pha dung dịch chuẩn như sau :
(a) Dung dịch chuẩn đồng
Hoà tan 3,928 g sunfat đồng CuSO4.5 H2O tính khiết trong nước, dùng nước pha loãng tới 1.000 ml ở nhiệt độ 20°, trong bình định mức. Dùng nước pha loãng 10 ml này tới 100 ml trong bình định mức khi cần đến.
1 ml = 100 μg Cu
(b) Dung dịch chuẩn kẽm
Hoà tan 1,000 g bột kẽm tinh khiết trong một hỗn hợp gồm 10 ml nước và 5 ml axit clohydric [thuốc thử đặc biệt (d)] và dùng nước pha loãng tới 1000 ml ở nhiệt độ 20°, trong bình định mức. Dùng nước pha loãng 10 ml dung dịch này tới 100 ml trong bình định mức khi cần.
1 ml = 100 μg Zn
(c) Dung dịch chuẩn crom
Dùng nước pha loãng 5.80 ml dung dịch natri dicromat tới 100 ml ở nhiệt độ 20° trong bình định mức, khi cần.
1 ml = 100 μ Cr
(d) Dung dịch chuẩn antimoan
Hoà tan 2,668 g kali antimoan tactrac K (SbO) C4H4O6 trong nước cất, dùng nước pha loãng tới 1000
ml ở nhiệt độ 20°, trong bình định mức. Dùng nước pha loãng 10,0 ml này tới 100 ml trong bình định mức, khi cần.
1 ml = 100 μ Sb
(e) Dung dịch chuẩn chì
Hoà tan 1,60 g nitrat chì Pb(NO3)2 trong axit nitric (10 ml axit nitric đặc pha loãng với 20 ml nước, đã đun sôi để loại bỏ khói nitơ, và để nguội) và dùng nước pha loãng tới 1000 ml, trong bình định mức. Dùng nước pha loãng 10,0 ml dung dịch này tới 500 ml ở nhiệt độ 20° trong bình định mức, khi cần.
1 ml = 100 μPb
(f) Dung dịch chuẩn bari
Hoà tan 1,779 g bari clorua BaCl2.H2O trong nước cất, pha loãng bằng nước tới 1000 ml ở 20°, trong một bình định mức. Dùng nước pha loãng 10,0 ml dung dịch này tới 100 ml trong bình định mức một vạch, khi cần.
1 ml = 100 μBa
(g) Dung dịch chuẩn asen
Hoà tan 1,320 g oxit asen As2O3 bằng cách hâm nóng ở nhiệt độ không vượt quá 60° với 14 ml dung dịch hydroxit natri 5 N trong cốc 100 ml. Làm nguội, cho thêm 0,2 ml chỉ thị phenol phtalein và trung hoà bằng axit sunfuric 6 N. Chuyển dung dịch này vào bình định mức 1000 ml, có chứa 10 g natri hydro cacbonat được hoà tan trong nước, dùng nước rửa hết lượng trong cốc. Pha loãng với nước tới vạch mức ở nhiệt độ 20°, và trộn. Dùng nước pha loãng 5 ml dung dịch này tới 1000 ml ở 20° trong bình định mức, khi cần.
1 ml = 100 μ As
(h) Dung dịch chuẩn cadimi
Hoà tan 2,282 g 3CdSO4 . 8H20 trong nước cất, dùng nước pha loãng tới 1000 ml ở 20°, trong bình định mức. Dùng nước pha loãng 10,0 ml dung dịch này tới 500 ml ở 20° trong một bình định mức, khi cần.
1 ml = 20 μg Cd
8.6 Chuẩn bị dung dịch thử
Chuẩn bị dung dịch thử theo phương pháp I trong trường hợp các chất có thể hoà tan được trong axit loãng. Dùng phương pháp II đối với các chất khác.
8.6.1 Phương pháp I
Cân chính xác 2,5 g mẫu và hoà tan trong một hỗn hợp gồm 4 ml axit sunfuric và 5 ml axit clohydric. Chuyển dung dịch này vào một bình định mức 50 ml. Nếu đo bari từ dung dịch, thêm 0,0954 g natri clorua. Dùng nước pha loãng tới vạch. Gọi đó là dung dịch A.
8.6.2 Phương pháp II
Cân chính xác khoảng 2,5 g mẫu thử, đưa vào bình Kendal dung tích 100 ml - 150 ml và thêm 5 ml axit nitric loãng. Ngay khi phản ứng ban đầu lắng xuống, đun nhẹ cho đến khi các phản ứng mạnh hơn tiếp theo dừng và sau đó để nguội. Cho từ từ 4 ml axit sunfuric đậm đặc với tốc độ không gây sủi bọt nhiều khi đun (thường cần 5 phút - 10 phút) và sau đó đun nóng - cho đến khi chất lỏng có mầu thẫm rõ rệt, từ là bắt đầu hoá than.
Thêm từ từ axit nitric đậm đặc với từng lượng nhỏ, đốt nóng giữa mỗi lần thêm cho đến khi mầu thẫm lại. Không đốt quá mạnh để xẩy ra quá trình hoá than mạnh, hay asen bị mất đi; cần có một lượng nhỏ nhưng không quá dư axit nitric tự do có mặt trong suốt quá trình. Tiếp tục việc xử lý này đến khi dung dịch chỉ còn mầu vàng nhạt và không thẫm lại khi đun thêm. Nếu dung dịch vẫn còn có mầu, thêm 0,5 ml dung dịch axit pecloric và ít axit nitric đậm đặc và đun nóng khoảng 15 phút, sau đó thêm 0,5 ml dung dịch axit pecloric và đun thêm vài phút nữa.
Ghi lại tổng lượng axit nitric đậm đặc đã sử dụng. để nguội một chút và pha loãng với 10 ml nước. Dung dịch này cần phải hoàn toàn không có màu (nếu có nhiều sắt, nó có thể có màu vàng nhẹ). Đun sôi nhẹ, chú ý tránh trào ra ngoài, đến khi xuất hiện khói trắng. Để nguội, thêm 5 ml nước nữa và lại đun sôi nhẹ đến khi bốc khói. Cuối cùng để nguội, thêm 10 ml axit clohydric 5 N vào và đun sôi nhẹ ít phút. Để nguội và cho vào một bình định mức 50 ml, rửa sạch bình Kendal bằng những lượng nước nhỏ. Cho nước rửa này vào bình định mức đó và pha loãng với nước cho tới vạch mức. Nếu muốn đo lượng bari trong dung dịch, trước khi pha loãng, thêm 0,0954 g clorua kali làm chất đệm ion hoá để ngăn chặn việc ion hoá của bari. Gọi dung dịch này là dung dịch A.
Chuẩn bị một mẫu trắng với cùng lượng thuốc thử như đã dùng trong việc oxi hoá mẫu.
8.7 Xác định hàm lượng antimoan, bari, cadimi, crom, đồng. chì và kẽm bằng phương pháp hấp thụ nguyên tử
8.7.1 Chuẩn bị các dung dịch dựng đường cong chuẩn
Dùng các ống hút cho vào một loạt các bình định mức có dung tích 100 ml, 1, 2, 3, 4 và 5 ml các dung dịch chuẩn thích hợp [các chuẩn từ (a) tới (f) và (h)] rồi pha loãng khoảng 50 ml. Thêm 8 ml axit sunfuric đậm đặc [thuốc thử (c)] và 10 ml axit clohydric đậm đặc [thuốc thử (d)]. Lắc để hoà tan chúng. Đối với nguyên tố bari [chuẩn (f)], thêm vào 0,191 g clorua kali như một chất đệm ion hoá. Khi điều chế xong dung dịch, pha loãng dung dịch tới vạch bằng nước không có thành phần kim loại.
Khi đó các dung dịch này có hàm lượng 0; 1,0; 2,0; 3,0; 4,0; và 5,0 μg/ml các nguyên tố bari hoặc là đồng, kẽm, crom, hoặc antimoan, hay 0; 0,2; 0,4; 0,6; 0,8; và 1,0 μg/ml cadimi hoặc chì.
8.7.2 Điều kiện thiết bị
Chọn bước sóng và các loại khí sử dụng cho mỗi nguyên tố theo bảng sau :
| Nguyên tố | Bước sóng (nm) | Khí |
| Antimoan | 217,6 | Không khí / axetilen |
| Bari | 553,6 | Oxit nitơ / axetilen |
| Cadimi | 228,8 | Không khí / axetilen |
| Crom | 357,9 | Oxit nitơ / axetilen |
| Đồng | 324,8 | Không khí / axetilen |
| Chì | 283,3 | Không khí / axetilen |
| Kẽm | 213,9 | Không khí / axetilen |
Chế độ đặt các thông số của thiết bị khác nhay thay đổi tuỳ theo từng môđen, và một vài thông số nhất định đòi hỏi sự tối ưu hoá tại thời điểm sử dụng để đạt được kết quả tốt nháat. Vì vậy, các thiết bị thử, cần được điều chỉnh như mô tả trong các chỉ dẫn của nhà sản xuất, khi sử dụng kiểu ngọn lửa và đặt bước sóng đặc trưng như xác định ở trên.
8.7.3 Cách tiến hành
Đặt máy quang phổ hấp thụ nguyên tử ở những điều kiện thích hợp. Hút dung dịch chuẩn đậm đặc chứa nguyên tố cần xác định và tối ưu hoá các thông số thiết bị để cho thang đo rộng rãi hoặc có độ uốn tối đa trên thiết bị ghi biểu đồ. Đo độ hấp thụ của các dung dịch chuẩn khác và vẽ đồ thị thể hiện độ hấp thụ thực so với nồng độ của nguyên tố trong các dung dịch chuẩn. Hút dung dịch A thu được do hoà tan hoặc oxi hoá ướt mẫu thử và dung dịch đối chứng tương ứng rồi xác định độ hấp thụ thực. Sử dụng đồ thị đã vẽ ở trên để xác định nồng độ của nguyên tố trong dung dịch mẫu.
| Nồng độ của nguyên tố (μg/ml) x 50 | = mg/kg nguyên tố trong mẫu |
| Lượng mẫu cân (g) |
8.8 Xác hàm lượng asen và antimoan bằng kỹ thuật hấp thụ nguyên tử hydrua Asen và antimoan được xác định sau khi xử lý các hydrua dễ bay hơi của chúng đã thu được hoặc ngay từ các bình tạo ra nó hoặc, trong một vài dạng thiết kế, trong một bóng cao su được gắn với bình tạo khí. Sau đó các khí này được tách ra cùng với khí argon đưa vào ngọn lửa hydro.
8.8.1 Pha các dung dịch dựng đường cong chuẩn
Sử dụng một buret để cho vào một loạt các bình định mức, dung tích 100 ml 1, 2, 3, 4 và 5 ml dung dịch asen hoặc antimoan chuẩn [các chuẩn (g) và (d)] rồi pha loãng tới khoảng 50 ml với nước cất. Thêm 8 ml axit sunfuric đặc [thuốc thử (c)] và 10 ml axit clohydric [thuốc thử (d)]. Lắc để hoà tan. Khi pha xong dung dịch, pha loãng tới vạch mức bằng nước cất.
8.8.2 Điều kiện thiết bị
Sử dụng một máy quang phổ hấp thụ nguyên tử với catốt rỗng thích hợp hoặc một đèn xả không cực, chọn bước sóng cho asen (193,7 nm) hoặc ăng ti moan (217,6 nm).
8.8.3 Cách tiến hành
Đong 5,0 ml dung dịch chuẩn đậm đặc nhất cho vào một bình tạo phản ứng, thêm 25 ml nước và 2 ml axit clohydric 5 N [thuốc thử (e)]. Nút bình lại và loại hết không khí ra như mô tả trong hướng dẫn sử dụng, nạp đầy khí argon vào thiết bị. Ngăn cách bình phản ứng với bình nguyên tử hoá bằng van nhánh. Nhấc bình nguyên tử hoá ra và nhanh chóng thêm 1 viên natriborohydrua nặng khoảng 0,2 g [thuốc thử (h)] và nút lại.
Đảm bảo rằng tất cả những chỗ nối đều bảo đảm.
Khi phản ứng diễn ra chậm (20 - 30 giây) mở các vòi thích hợp để khí argon đẩy hydrua được tạo ra vào ngọn lửa. Khi đã loại bỏ toàn bộ hydrua như thiết bị ghi đã thể hiện, trả các khoá về vị trí ban đầu và làm sạch bình.
Tối ưu hoá các thông số thiết bị để có phạm vi đo tối đa cho chuẩn mạnh (đậm đặc nhất). Đo các chuẩn khác, dung dịch mẫu và dung dịch đối chứng theo cùng một trình tự tiến hành.
Dựng một đồ thị tương quan giữa chiều cao của Pic ghi được trên thiết bị ghi, với nồng độ asen và antimoan trong các dung dịch chuẩn. Dùng độ hấp thụ thực của mẫu chuẩn, ta sẽ đọc nồng độ của asen hoặc antimoan trong dung dịch từ đồ thị.
8.8.4 Tính toán
| Nồng độ asen hoặc antimoan (μg/ml) x 50 | = mg/kg asen hoặc antimoan trong mẫu thử |
| Lượng mẫu cân (g) |
8.9 Xác định thuỷ ngân bằng kỹ thuật hoá hơi hấp thụ nguyên tử
8.9.1 Nguyên tắc
Mẫu được tro hoá bằng cách đốt trong điều kiện hồi lưu với axit sunfuric và axit nitric. Hoàn thiện việc oxi hoá bằng cách thêm dung dịch permanganat kali. Sau khi thêm liên tiếp dung dịch hydroxylamin hydroclorua và dung dịch clorua thiếc, hàm lượng thuỷ ngân được xác định bằng thiết bị quang phổ hấp thụ nguyên tử hoá hơi lạnh.
8.9.2 Thuốc thử và dụng cụ
+ Các thuốc thử đặc biệt
(a) Axit nitric, khối lượng riêng 1.40;
(b) Axit sunfuric, khối lượng riêng 1.84;
(c) Axit sunfuric, xấp xỉ 3,5 M. Điều chế bằng cách pha một thể tích axit sunfuric đậm đặc (b), với 4 thể
thích nước;
(d) Axit sunfuric, xấp xỉ 1 M. Điều chế bằng cách pha một thể tích axit sunfuric 3,5 M (c), với 2,5 thể
thích nước;
(e) Axit clogydric, khối lượng riêng 1,18;
(f) Dung dịch permanganat kali, 50,0 g/L;
(g) Dung dịch hydroxylamin hydroclorua, 100,0 g/L;
(h) Dung dịch clorua thiếc. Điều chế bằng cách hoà tan 25,0 g clorua thiếc (SnCl2.2H2O) trong 50 ml axit clohydric (e). Phe với nước thành 250 ml và cho sục khí nitơ qua dung dịch. Bảo quản với một vài hạt thiếc kim loại;
(i) Hỗn hợp axit crom. Hoà tan 4,0 g kali dicromat trong 300 ml axit sunfuric 3,5 M (c) và pha với nước thành 1 lít;
(j) Mangan peclorat, dạng hạt, để làm khô khí;
(k) Clorua thuỷ ngân.
+ Các dung dịch chuẩn
Dùng các dung dịch chuẩn có sẵn trên thị trường, hoặc, điều chế các dung dịch chuẩn như sau :
(a) Dung dịch clorua thuỷ ngân, 0,5 mg Hg/ml;
Cân 0,677 g clorua thuỷ ngân (k) chính xác đến 0,1 mg, hoà tan trong khoảng 250 ml axit sunfuric 3,5 M
(c) trong một bình định mức có dung tích 1 lít, thêm khoảng 700 ml nước, sau đó nhỏ từng giọt dung dịch permanganat kali (f) cho đến khi bền mầu sắc. Thêm nước cho tới vạch mức và trộn kỹ. Pha mới dung dịch này 3 tháng một lần.
(b) Dung dịch clorua thuỷ ngân, 0,02 μg Hg/ml.
Pha loãng dung dịch chuẩn clorua thuỷ ngân 0,5 mg Hg/ml [chuẩn (A)] tới một hệ số 25.000 bằng cách pha loãng liên tiếp với axit sunfuric [thuốc thử đặc biệt (d)], ví dụ, 10 ml pha thành 250 ml hai lần, rồi lại pha 10 ml thành 400 ml. Trước khi làm đầy tới vạch dung dịch pha loãng cuối cùng, thêm dung dịch permanganat kali [thuốc thử đặc biệt (f)] nhỏ từng giọt cho tới khi màu sắc không đổi. Pha mới dung dịch này hàng ngày.
+ Thiết bị thử
 Tất cả các dụng cụ thuỷ tinh phải được làm sạch bằng axit nitric nóng [thuốc thử đặc biệt (a)] và rửa kỹ bằng nước trước khi sử dụng.
Tất cả các dụng cụ thuỷ tinh phải được làm sạch bằng axit nitric nóng [thuốc thử đặc biệt (a)] và rửa kỹ bằng nước trước khi sử dụng.
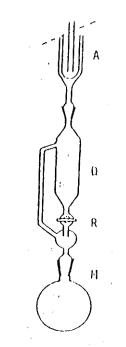
- Bình vô cơ hoá được lắp với bình ngưng hồi lưu (xem hình 2).
- Các bình cầu, có một nút thuỷ tinh nối với hai ống, để kéo hơi thuỷ ngân và có một vạch đo đánh dấu yêu cầu khi đo.
Dung tích của bình tròn và vị trí vạch đo tuỳ thuộc vào máy quang phổ hấp thụ nguyên tử được sử dungj.
Làm sạch bình tròn lần lượt bằng hỗn hợp axit crom [thuốc thử đặc biệt (i)], nước máy và nước cất hai lần trước khi dùng.
- Thiết bị hấp thụ hơi nước, chứa peclorat mangan [thuốc thử đặc biệt (j)].
- Máy quang phổ hấp thụ nguyên tử, thích hợp cho việc xác định hơi thuỷ ngân lạnh trong mạch kín hoặc hở, có thiết bị ghi.
8.9.3 Cách tiến hành
8.9.3.1 Tro hoá
Cân chính xác đến 2 mg khoảng 0,5 g mẫu chứa không quá 0,5 μg tổng lượng thuỷ ngân. Cho mẫu vào bình phản ứng (M), và thêm một vài hạt thuỷ tinh. Nối bình phản ứng với ống ngưng (D) và đóng van (R). (Hình 2).
![]() Đưa vào ống ngưng 25 ml axit nitric [thuốc thử đặc biệt (a)] rồi cho thêm 10 ml axit sunfuric [thuốc thử đặc biệt (b)]. Lắp thiết bị và mở bình ngưng tụ (A). Mở khoá cẩn thận và để cho những lượng nhỏ hỗn hợp các axit chạy vào bình phản ứng. Ngắt dòng axit nếu thấy phản ứng xảy ra quá mãnh liệt.
Đưa vào ống ngưng 25 ml axit nitric [thuốc thử đặc biệt (a)] rồi cho thêm 10 ml axit sunfuric [thuốc thử đặc biệt (b)]. Lắp thiết bị và mở bình ngưng tụ (A). Mở khoá cẩn thận và để cho những lượng nhỏ hỗn hợp các axit chạy vào bình phản ứng. Ngắt dòng axit nếu thấy phản ứng xảy ra quá mãnh liệt.
Tháo hết các chất trong ống ngưng vào bình phản ứng, trộn kỹ các thành phần trong bình này bằng cách lắc cẩn thận và để khoá ở vị trí mở.
Đốt nóng bình phản ứng từ từ. Ngay khi hết nổi bọt, đóng khoá (R), Tiếp tục đun để thu lấy chất ngưng tụ trong ống ngưng.
Ngừng đun khi các chất trong binh phản ứng bắt đầu hoá than. Cho một lượng nhỏ chất ngưng tụ vào bình phản ứng, lại đóng khoá và tiếp tục đun bình phản ứng. Lập lại trình tự này cho đến khi các chất trong bình đã hoá than trong khi đun.
Khi hiện tượng hoá than đã dừng, đun và thêm chất ngưng tụ vào ngay khi có khói trắng. Tiếp tục đun và thêm chất ngưng tụ như trên trong 1 giờ. Cuối cùng, đốt các chất trong bình cho đến khi thấy khói trắng.
Ngừng đun và để nguội cho tới xấp xỉ 40°. Mở van để cho tất cả các chất ngưng tụ đi vào bình phản ứng. Rửa thiết bị từ đỉnh bình ngưng tụ bằng 5 ml - 10 ml nước thu lấy nước rửa vào bình phản ứng và tháo nó ra khỏi khoang ống ngưng.
8.9.3.2 Xử lý dung dịch thử
Nhỏ từng giọt dung dịch permanganat kali [thuốc thử đặc biệt (f)] vào bình phản ứng, khuấy cho đến khi có màu hồng bền. Ghi lại lượng thuốc (f) đã được sử dụng, (nếu lượng dung dịch này vượt quá 10 ml, hãy lặp lại quy trình tro hoá như trên).
Đun từ từ đến sôi, rồi để nguội.
Rót lượng chứa trong bình phản ứng vào một bình cầu, rửa bình phản ứng bằng nước và cho dịch rửa này vào bình cầu trên.
Đo hàm lượng thuỷ ngân (xem bên dưới) trong cùng ngày xử lý dung dịch thử.
8.9.3.3 Đo hàm lượng thuỷ ngân
Cho 5 ml hydroxylamin hydro clorua [thuốc thử đặc biệt (g)] vào bình tròn nói trên và làm đầy tới vạch bằng nước cất hai lần, hoặc axit sunfuric [thuốc thử đặc biệt (d)] trong trường hợp các dung dịch chuẩn. Thêm 5 ml dung dịch clorua kẽm [thuốc thử đặc biệt (g)] vào, lắp bình cầu, nối nó với thiết bị hấp thụ hơi và với máy quang phổ hấp thụ nguyên tử. Đưa máy quang phổ vào hoạt động.
Trộn kĩ các chất trong bình cầu bằng cách lắc nhẹ, đưa không khí hoặc khí nitơ đi qua, đo và ghi lại. Tiến hành đo càng nhanh càng tốt ngay sau khi thêm clorua kẽm. Nếu sử dụng hệ thống hở, hãy đợi 30 giây trước khi cho không khí hoặc khí nitơ đi qua.
8.9.3.4 Đồ thị chuẩn
Cho lần lượt 2, 5, 10, 15 và 25 ml dung dịch thuỷ ngân chuẩn [tiêu chuẩn (b)] vào 5 bình cầu khác nhau và 25 ml axit sunfuric [thuốc thử đặc biệt (d)] vào một bình cầu thứ sáu. Nhỏ thêm dung dịch permanganat kali [thuốc thử đặc biệt (f)] vào và lắc các bình, cho đến khi không có sự biến đổi về màu sắc.
Đo hàm lượng thuỷ ngân như đã mô tả ở trên.
Dựng đồ thị chuẩn, với các giá trị hấp thụ đo được là các tung độ và các hàm lượng thuỷ ngân tương ứng tính bằng μg là hoành độ. Các dung dịch chuẩn làm việc có chứa lần lượt : 0, 0,04; 0,1; 0,2; 0,3; và 0,5 μg thuỷ ngân tương ứng.
8.9.3.5 Phương pháp bổ sung
Phương pháp bổ sung này có thể được dùng nêu sử dụng một hệ thống hở.
Đưa một trong số dung dịch chuẩn làm việc (xem mục 8.9.3.4) vào một bình cầu và thêm một lượng nước của dung dịch mẫu thu được sau khi xử lý (xem mục 8.9.3.2). Lượng thuỷ ngân trong bình phải nằm trong dải mà máy đo thể hiện một quan hệ tuyến tính. Đo hàm lượng thuỷ ngân như mô tả trong mục 8.9.3.3. Nếu cần thiết, tiến hành xác định vài lần và dùng các dung dịch chuẩn khác nhau.
8.9.3.6 Xác định mẫu đối chứng (mẫu trắng)
Tiến hành tất cả các bước, từ tạo tro đến đo đạc, trừ việc đưa mẫu vào. Khi xử lý dung dịch, thêm một lượng dung dịch permanganat kali [thuốc thử đặc biệt (f)] bằng với lượng đã dùng cho mẫu thí nghiệm.
8.9.4 Tính toán
Đọc từ đồ thị chuẩn các lượng thuỷ ngân, tính bằng μg, tương ứng với các giá trị hấp thụ đo được. Lấy lượng thủy ngân tìm được ở mẫu thử trừ đi lượng thuỷ ngân tìm thấy trong mẫu trắng.
| Hàm lượng thực của thuỷ ngân (μg) | = mg/kg Hg trong mẫu thử |
| Lượng mẫu cân (g) |
Trừ khi được hướng dẫn khác cho từng chuyên luận, phương pháp II được trình bày dưới đây được ưa dùng hơn phương pháp I.
9.1 Phương pháp I (quy trình Gutzeit)
9.1.1 Điều chế dung dịch mẫu
Dung dịch thu được bằng cách xử lý mẫu như đã hướng dẫn cho từng chuyên luận, được dùng trực tiếp làm dung dịch mẫu trong quy trình tiến hành.
9.1.2 Pha chế dung dịch chuẩn asen
Cho vào 50 ml nước : 10 ml thuốc thử axit clohydric nhiễm thiếc và 1.0 ml thuốc thử asen loãng. Dung dịch thu được này sau khi xử lý như mô tả trong quy trình dưới đây, sẽ tạo màu trên giấy clorua thuỷ ngân, được dùng làm màu chuẩn, tương đương với 10 μg asen.
9.1.3 Cách tiến hành
Chuyển dung dịch (D) vào một bình hình nón có dung tích 120 ml (xem hình 3). Bình này được lắp một nút cao su, bên trong có một ống thuỷ tinh dài 200 mm và đường kính trong của ống là 6.5 mm xuyên qua. Đầu dưới của ống thuỷ tinh được cắt vát một góc và có một lỗ đường kính không nhỏ hơn 2 mm ở thành (cạnh) của ống. Cách khoảng 40 mm về phía trên của nút, ống được cắt rới vuông góc thành hai phần : một đĩa bằng giấy thử (A) có đường kính bằng đường kính ngoài của ống được đặt xen vào giữa hai đoạn ống. Hai đoạn ống có giấy thử này được nối chặt với nhau bằng một ống cao su (B).
Giấy thử hình tròn được làm từ giấy lọc (Whatman No.1 hoặc tương đương), ngâm trong dung dịch clorua thuỷ ngân 5 % pha trong ethanol, và được làm khô nhờ một dòng khí.
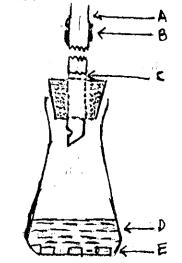 A. Đĩa thí nghiệm giấy thử clorua thuỷ ngân. B. Đoạn ống nối cao su.
A. Đĩa thí nghiệm giấy thử clorua thuỷ ngân. B. Đoạn ống nối cao su.
C. Gạc bông được bão hoà với axetat chì. D. Dung dịch thử.
E. Các miếng nhôm vuông
Hình 3 - Thiết bị để thử giới hạn asen
Nút lỏng lẻo đầu dưới của ống bằng gạc bông ngâm trong dung dịch axetat chì 5 %, và làm khô (C).
Trong bình nón, bỏ thêm ba miếng lá nhôm vuông (8 mm x 8 mm x 1 mm) (E) và nút chặt bình ngày bằng một nút cao su. Đặt bình ở trong bể nước ở nhiệt độ 25° trong 45 phút.
Đồng thời, tiến hành một thử nghiệm song song, dùng dung dịch asen chuẩn thay cho mẫu thử. So sánh màu sắc của hai miếng giấy thử clorua thuỷ ngân. Cường độ màu từ mẫu thử không được lớn hơn cường độ màu của màu chuẩn.
9.2 Phương pháp II (quy trình đo màu)
9.2.1 Thiết bị và dụng cụ
Thiết bị thí nghiệm thông thường được trình bày trong sơ đồ kèm theo. Nó bao gồm một bình sinh asen 125 ml (A), với một cổ nối tiêu chuẩn 24/40 (B) lắp vừa khít với bộ phận lọc hơi (C) và một ống hấp thụ (E) nối bởi một ống mao dẫn có đường kính trong 2 mm, đường kính ngoài 8 mm qua một ổ nối hình cầu lõm (D), được giữ chặt bằng kẹp số 12, nối với các bộ phận. Nói cách khác, một thiết bị thể hiện nguyên tắc của tổ hợp chung như đã được mô tả là có thể được sử dụng.
Hình 4 - Thiết bị để thử giới hạn asen - Phương pháp II
9.2.2 Thuốc thử
9.2.2.1 Dung dịch bạc diethyldithiocarbamat
Hoà tan 1 g bạc dietyldithiocarbamat đã tái kết tinh, (C2H5)2NCSSAg trong 200 ml pyridin (cấp thuốc thử) trong một tủ hút. Bảo quản dung dịch này trong một lọ màu sẫm và dùng trong vòng một tháng.
Bạc dietyldithiocarbamat sẵn có trên thị trường hoặc được điều chế như sau : Hoà tan 1.7 g nitrat bạc (cấp thuốc thử) trong 100 ml nước. Trong một bình khác, hoà tan 2,3 g natri dietyldithiocarbamat (C2H5)2NCSSNa.3H2O, trong 100 ml nước, và lọc. Làm nguội cả hai dung dịch đến khoảng 15°, trộn lẫn hai dung dịch này. Trong khi khuấy, thu chất kết tủa màu vàng trong một chén nung, hoặc phễu thuỷ tinh xốp trung bình và rửa với khoảng 200 ml nước lạnh.
Kết tinh lại thuốc thử, hoặc ở dạng điều chế như chỉ dẫn ở trên, hoặc dạng mua sẵn, theo cách sau : hoà tan trong pyridin mới dc chưng cất, dùng khoảng 100 ml dung môi cho mỗi g thuốc thử, và lọc. Thêm vào một thể tích nước lạnh bằng thể tích dung dịch pyridin, trong khi khuấy, Lọc lấy ra kết tủa sử dụng lực hút, rửa bằng nước lạnh và làm khô trong chân không ở nhiệt độ phòng trong 2 đến 3 giờ. Muối khô có màu vàng tinh khiết, và sẽ phải không biến đổi đặc tính sau một tháng khi được bảo quản trong lọ sẫm màu. Loại bỏ bất kỳ chất nào biến đổi màu hoặc bốc mùi hắc, nặng.
9.2.2.2 Dung dịch chuẩn asen
Cân chính xác 132,0 mg trioxit asen đã được tán thành bột mịn và được làm khô trong 24 giờ ở bên trên một chất có tác dụng hút ẩm, làm khô thích hợp, và hoà tan nó trong 5 ml dung dịch natri hydroxit (1 : 5). Trung hoà dung dịch với axit sunfuric loãng, cho thêm 10 ml dư, và pha loãng thành 1000 ml bằng nước mới đun sôi và trộn. Chuyển 10 ml dung dịch này vào một bình định mức có thể tích 1000 ml, thêm 10 ml axit sunfuric loãng, pha loãng tiếp tới thể tích bằng nước mới đun sôi và trộn.
Chỉ sử dụng dung dịch cuối cùng này, có chứa 1 μg asen (As) trong mỗi ml, trong vòng 3 ngày.
9.2.2.3 Dung dịch clorua thiếc
Hoà tan 40 g dihydrat clorua thiếc (ở cấp thuốc thử) SnCl2.2H2O trong 100 ml axit clohydric. Bảo quản dung dịch này trong một lọ thuỷ tinh và sử dụng trong vòng 3 tháng.
9.2.2.4 Bông tẩm axetat chì
Ngâm bông vào dung dịch bão hoà axetat chì (cấp thuốc thử), gạn ép hết dung dịch thừa và làm khô trong chân không ở nhiệt độ phòng.
Chú thích - Khi điều chế và sử dụng bông này cần chú ý tránh bị nhiễm chì.
9.2.3 Xử lý dung dịch mẫu
Dung dịch thu được bằng cách xử lý mẫu như đã chỉ dẫn trong một chuyên luận cụ thể, sẽ được dùng trực tiếp như một dung dịch mẫu trong tiến trình thử nghiệm. Dung dịch mẫu từ các hợp chất hữu cơ được xử lý trong bình phát khí (A) theo trình tự chung sau đây, nếu không có hướng dẫn khác :
Cảnh báo : Một vài chất có thể phản ứng bất ngờ, cho nổ lớn khi được vô cơ hoá hydro peroxyt. Những biện pháp phòng ngừa an toàn phù hợp phải được áp dụng luôn luôn.
Chú thích - Nếu có mặt các hợp chất chứa halogen, hãy dùng nhiệt độ thấp hơn khi làm nóng mẫu với axit sunfuric, không đun sôi hỗn hợp, và cẩn thận cho thêm peroxyt trước khi bắt đầu than hoá, để ngăn ngừa mất mát asen hoá trị ba.
Cho 1 g mẫu vào trong bình phát khí, thêm 5 ml axit sunfuric và một vài hạt thuỷ tinh và đem vô cơ hoá ở nhiệt độ không quá 120° trên một bếp điện trong một cái tủ hút cho đếnk hi bắt đầu hoá thành than (một vài mẫu có thể càan cho thêm một lượng axit sunfuric để làm ẩm hoàn toàn, nhưng tổng lượng thêm này không nên quá 10 ml). Sau khi mẫu đã bắt đầu được phân huỷ bằng axit, cẩn thận cho vào từng giọt hydro peroxyt 30 % đợi cho phản ứng dịu đi và đốt nóng lại sau các giọt cho thêm.
Vai giọt đầu tiên phải được thêm vào rất chậm và trộn kỹ để tránh phản ứng quá nhanh, và ngừng đun nóng nếu sủi bọt quá nhiều. Khuấy dung dịch trong bình để ngăn ngừa các chất không bị phản ứng bị cháy kết ở thành hoặc đáy bình trong khi vô cơ hoá. Duy trì điều kiện oxy hoá trong quá trình vô cơ hoá bằng cách thêm một lượng nhỏ peroxyt bất cứ lúc nào hoõn hợp có màu nâu hoặc màu sẫm.
Tiếp tục vô cơ hoá cho đến khi chất hữu cơ bị phân huỷ, tăng từ từ nhiệt độ của bếp đốt đến 250° ữ 300° cho đến khi hơi khói axit sunfuric bốc ra nhiều và dung dịch trở nên không màu, hoặc chỉ còn màu vàng rơm nhạt.
Làm nguội, cẩn thận cho thêm 10 ml, tiếp tục cho bay hơi (khói axit sunfuric bốc lên) và làm nguội. Thêm một cách thận trọng 10 ml nước, trộn, rửa thành bình bằng vài ml nước, và pha loãng tới 35 ml.
9.2.4 Cách tiến hành
Nếu dung dịch mẫu không được xử lý trong bình phát khí, cho vào bình một thể tích dung dịch, được điều chế như đã chỉ dẫn, tương đương với 1 g chất đang được thử và thêm nước để tạo nên 35 ml.
Thêm 20 ml axit sunfuric loãng (1 : 5) 2 ml kali iodua và 0,5 ml dung dịch clorua thiếc, trộn lẫn. Để yên hỗn hợp trong 30 phút ở nhiệt độ phòng. Nhồi vào ống lọc hơi (C) 2 nút bông tẩm axetat chì, để một khoảng không khí nhỏ giữa 2 nút, bôi trơn chỗ nối (B) và (D) bằng mỡ chuyên dụng nếu cần thiết, và nối bộ lọc hơi đốt với ống hấp thụ (E). Chuyển 3 ml dung dịch bạc dietyldithiocarbamat tới ống hấp thụ, thêm 3 g hạt kẽm (20 mesh) vào hỗn hợp trong bình và nút ngay ống nối có cổ chuẩn vào bình, để hydrro phát ra và sự biến đổi màu diễn ra ở nhiệt độ trong phòng (25° ± 3°) trong 45 phút, xoay nhẹ bình cách các quãng khoảng 10 phút / lần. (Việc cho thêm một lượng nhỏ isopropanol vào bình có thể cải thiện tính đồng đều của tốc độ phát khí), tháo ống hấp thụ khỏi bộ phận lọc hơi và máy phát khí và cho dung dịch bạc dietyldithiocarbamat vào cuvet 1 cm để đo.
Xác định độ hấp thụ ở bước sóng hấp thụ lớn nhất giữa 535 nm và 540 nm với một máy đo phổ hoặc đo màu thích hợp, dùng dung dịch bạc dietyldithiocarbamat như là mẫu trắng.
Độ hấp thu của bất kỳ màu đỏ nào từ dung dịch mẫu không được đậm hơn độ hấp thu của 3 ml dung dịch asen chuẩn khi được xử lý theo cách giống nhau và dưới các điều kiện giống như mẫu. Nhiệt độ trong phòng trong quá trình phát asen từ dung dịch chuẩn nên giữ trong khoảng ± 2° với nhiệt độ khi xác định mẫu.
Chú thích 1 - Các kim loại hoặc muối của các kim loại như crom, côban, đồng, thuỷ ngân, môlípđen, niken, paladi và bạc được cho là gây trở ngại cho việc sinh asen. Antimoan mà nó tạo ra stibin là kim loại duy nhất dường như tạo ra một nhiễu dương tính về màu sắc với bạc diethyl dithocacbamat. Stibin sẽ cho màu đỏ có độ hấp thụ cực đại ở 510 nm, nhưng ở 535 - 540 nm, độ hấp thụ của phức hợp antimoan lại giảm tới mức các kết quả xác định sẽ không chênh lệch đáng kể.
Chú thích 2 - Tất cả các thuốc thử dùng trong phép thử giới hạn asen cần có lượng asen rất thấp.
Phép thử giới hạn được mô tả dưới đây nhằm xác định xem liệu mẫu thử chứa crôm nhiều hay ít hơn 20 mg/kg.
10.1 Cách tiến hành
Cân 1,0 g mẫu thử vào một đĩa thạch anh. Đốt thành than, nâng dần nhiệt độ. để nguội, cho 10 ml dung dịch magie nitrat 25 %; cho bay hơi, làm nóng dần cho đến khi không còn hơi nitơ thoát ra. Nung mẫu trong lò ở 600°C cho đến khi tất cả các hạt đen biến mất (1h).
Hoà tan cặn với 10 ml axit sunfuric 4 N và 20 ml nước. Đun nóng trên bếp cách thuỷ khoảng 5 phút.
Thêm 0,5 ml pecmanganat kali 0,1 N. Thêm nhiều hơn, nếu dung dịch nhạt màu. Đậy bằng 1 mặt kính đồng hồ, và đun nóng trên bếp cách thuỷ khoảng 20 phút. Cho thêm dung dịch azid natri 5 %, cứ 10 giây cho một giọt, cho tới khi thuốc tím dư đã được loại hết (tránh thừa azid natri; thường thì hai giọt là đủ). Làm nguội dung dịch trong vòi nước chảy và lọc nếu có dioxít mangan. Cho dung dịch vào một bình định mức 50 ml. Thêm vào 2,5 ml dihydrogenphosphat natri 5 M, thêm 2 ml diphenyl carbazide rồi đổ đầy nước tới vạch. Đo độ hấp thụ ở 540 nm sau khi cho diphenyl carbazide vào 30 phút. Một mẫu trắng với hai thuốc thử như trên sẽ cho thấy không có màu hoặc chỉ có màu tía nhẹ.
Làm một thử nghiệm song song đồng thời, với 1,00 ml crom chuẩn (1 ml = 20 μg crôm) và 1 vài ml đường sacaz được đặt vào một đĩa thạch anh khác. Xử lý hỗn hợp này đúng như mẫu và đo độ tắt ở cùng bước sóng. Tính toán lượng crôm của mẫu từ 2 giá trị độ tắt đã quan sát được.
11 Phép thử giới hạn tổng kim loại nặng
Đây là một phép thử theo kinh nghiệm “bắt - tất“ đối với các kim loại nặng dưới đây, (ngoài chì) : thuỷ ngân, cadimi, antimoan, asen (một phần), bạc, đồng và một vài kim loại khác. Tất cả các kim loại này tạo màu với hydrogen sunfua và thí nghiệm được thiết kế để chứng tỏ rằng tổng lượng có mặt, được biểu diễn bằng chì, không vượt quá các giới hạn kim loại nặng đã được xác định trong nhiều chuyên luận khác nhau. Kẽm và thiếc cũng tạo thành sunfit và mặc dù chúng không bị ngả màu ở độ pH của thí nghiệm (3 - 4) chúng có thể ảnh hưởng tới phép thử ở một mức độ nào đó.
Phép thử này là hữu ích để chỉ sự có mặt của tạp chất kim loại nặng trong một vật liệu thô từ một nguồn mới, và phát hiện sự nhiễm bẩn ngẫu nhiên mà nhà sản xuất có thể không nhận biết được. Nó cũng cung cấp sự xác nhận về “thực hành sản xuất tốt“.
Tuy nhiên nếu như phương pháp sản xuất hoặc cấp chất lượng vật liệu thô được dùng vì lý do để tin tưởng rằng một tạp cháat như thuỷ ngân hoặc cadimi có thể có mặt thì, một phép thửu cụ thể về tạp chất nghi ngờ cần được thực hiện.
Phương pháp I được dùng cho các chất mà chúng tạo ra các dung dịch không màu trong suốt trước khi thêm in sunfit vào, và cần dùng trong quy trình thử nghiệm trừ khi có hướng dẫn khác trong chuyên luận riêng. Phương pháp II được sử dụng cho các chất không sinh ra các dung dịch không màu trong suốt trong các điều kiện thử nêu trong phương pháp I hoặc cho những chất mà, do bản chất phức tạp của chúng, sẽ gây nhiễm với sự kết tủa của các kim loại nặng bởi sunfit.
11.1 Thuốc thử
a) Ammoni
Pha loãng 400 ml hydroxit amoni (cấp thuốc thử) tới 1000 ml với nước. b) Axít clohydric
Dùng axit clohydric (cấp thuốc thử) trong điều chế mọi dung dịch axit clohydric được sử dụng trong phép thử này.
c) Dung dịch nitrat chì
Hoà tan 159,8 mg nitrat chì, Pb(NO3) 2, trong 100 ml nước có chứa 1 ml axit nitric, sau đó pha loãng với nước thành 1000 ml và trộn. Dung dịch này cần được pha chế và bảo quản trong một bình chứa thuỷ tinh không có các muối chì.
d) Dung dịch chì chuẩn
Vào ngày dùng, pha loãng 10,0 ml dung dịch gốc nitrat chì, được đo chính xác, với nước để có 100,0 ml. Mỗi ml dung dịch điều chế như thể sẽ chứa một lượng tương đương 10 μg ion chì (Pb).
11.2 Cách tiến hành
Chú thích - Trong các cách tiến hành dưới đây cho phương pháp I và phương pháp II, nếu không điều chỉnh được chính xác đọ pH của dung dịch trong các giới hạn đặc trưng có thể làm ảnh hưởng đến độ nhạy của phép thử một cách đáng kể.
11.2.1 Phương pháp I
a) Dung dịch A
Dùng pipet nhỏ 2 ml dung dịch chì chuẩn (20 μg ion chì (Pb) vào một ống Nessler dung tích 50 ml) trừ khi được chỉ dẫn khác trong chuyên khảo. Chỉnh độ pH từ 3,0 đến 4,0 (giấy chỉ thị pH phạm vị hẹp) bằng cách thêm axit axetic loãng hoặc amoniac, pha loãng với nước thành 40 ml và trộn.
b) Dung dịch B
Cho vào một ống Nessler dung tích 50 ml như đã dùng cho dung dịch A, 25 ml dung dịch mẫu đã được xử lý như hướng dẫn trong chuyên khảo riêng. Chỉnh độ pH đến khoảng 3,0 và 4,0 (giấy chỉ thị pH phạm vi hẹp) bằng cách thêm axit axetic loãng hoặc amoniac, pha loãng tới 40 ml với nước và trộn.
c) Dung dịch C
Cho vào một ống Nessler 50 ml thứ ba, như hai cái đã được dùng cho dung dịch A và B, 25 ml dung dịch mẫu đã được xử lý như hướng dẫn trong chuyên khảo và cho thêm một thể tích dung dịch chì chuẩn giống như đã cho đối với dung dịch A. Điều chỉnh độ pH đến khoảng 3,0 và 4,0 (giấy chỉ thị pH phạm vi hẹp) bằng cách thêm axit axetic loãng hoặc amoniac, pha loãng bằng nước tới 40 ml và trộn. Thao tác trong tủ hút (fume hood), thêm vào mỗi ống 10 ml thuốc thử hydrogen sunfit mới pha, trộn, để yên trong 5 phút và nhìn soi xuống trên một bề mặt trắng. Màu của dung dịch B không sẫm hơn màu của dung dịch A, và màu của dung dịch C giống như hoặc sẫm hơn màu của dung dịch A. Nếu màu của dung dịch C mà nhạt hơn màu của dung dịch A thì mẫu thử đang tạo ra hiện tượng nhiễu và phải dùng phương pháp II.
11.2.2 Phương pháp II
Tiến hành như đã hướng dẫn ở phương pháp I để điều chế dung dịch B như sau :
Thao tác trong một tủ hút (fume hood), cho một lượng xác định của mẫu, đã được cân chính xác, vào trong một chén nung thích hợp, cho axit sunfuric đủ để làm ướt mẫu, và đốt cẩn thận ở nhiệt độ thấp cho đến khi cháy hết, đậy hờ chén nung bằng một nắp thích hợp trong khi đốt. Sau khi chất thử cháy hết thành than, cho 2 ml axit nitric và 5 giọt axit sunfuric, và làm nóng cẩn thận cho tới khi khói trắng bốc ra, sau đó tiến hành đốt nóng, tốt nhất là ở trong lò nung, ở 500° tới 600° cho tới khi tất cả than cháy hết. Làm nguội, cho 4 ml axit clohydric loãng (1 : 2), đậy kín và vô cơ hoá trên thiết bị cách hơi nước 10 đến 15 phút. Mở nắp, và cho bốc hơi từ từ trên thiết bị cách hơi nước cho đến khi khô.
Làm ẩm cặn bằng 1 giọt axit clohydric, cho 10 ml nước nóng và để vô cơ hoá trong 2 phút. Cho vào từng giọt amoniac cho tới khi dung dịch vừa tới chớm kiềm trên giấy quỳ, pha loãng bằng nước tới 25 ml và điều chỉnh độ pH đến giữa 3,0 và 4,0 (giấy chỉ thị pH phạm vị hẹp) bằng cách cho thêm axit axetic loãng. Lọc nếu cần, rửa chén nung và phễu lọc bằng 10 ml nước, cho dung dịch này và nước rửa vào một ống Nessler 50 ml, pha loãng với nước tới 40 ml và trộn.
Pha chế dung dịch C theo cách tương tự như đối với dung dịch B, thêm vào mẫu trong chén nung, một thể tích dung dịch chì chuẩn giống như đã cho vào dung dịch A. Tiến hành như ppp I từ “theo thao tác trong tủ hút, thêm... “.
Lấy 0,5 g mẫu cân chính xác tới mg, cho thêm 2 ml axit clohydric và cho bay hơi đến khô trên thiết bị cách hơi nước. Hoà tan cặn trong 2 ml axit clohydric và 20 ml nước, và cho thêm một vài giọt brom (Br). Đun sôi dung dịch trong tủ hút (fume hood) để đuổi brom (Br), làm nguội, pha loãng với nước, tới 25 ml. Sau đó cho thêm 50 mg amonium persunfat và 5 ml amoni thioxyanat. Bất kỳ màu đỏ nào được tạo ra không được đậm hơn màu của dung dịch đối chứng được thực hiện cách giống như dung dịch thử nhưng có chứa một lượng sắt chuẩn, như đã nêu trong chuyên luận riêng, thay cho dung dịch mẫu.
13.1 Thuốc thử đặc biệt
Lựa chọn các thuốc thử có hàm lượng chì thực tế càng thấp càng tốt, và phải bảo quản tất cả các dung dịch trong các lọ thuỷ tinh borosilicat, được tráng kỹ tất cả các lọ thuỷ tinh bằng axit nitric loãng ấm (1 : 2) tiếp theo rửa bằng nước.
a) Dung dịch xyanua - amoniac (amoni - cyanide)
Hoà tan 2 g kali xyanua trong 15ml thuốc thử amoniac đậm đặc và pha loãng với nước tới 100 ml.
b) Dung dịch chuẩn chì pha loãng (1 μg Pb trong 1 ml)
Ngay trước khi dùng, chuyển 10 ml chuẩn chì có chứa 10 μg chì/1 ml vào bình định mức 100 ml, pha loãng đến thể tích bình bằng axit nitric loãng (1 : 100) và trộn.
13.2 Dung dịch mẫu
Dung dịch thu được bằng cách xử lý mẫu như chỉ dẫn trong chuyên luận riêng sẽ được sử dụng trực tiếp làm dung dịch mẫu trong quy trình thử nghiệm. Các dung dịch mẫu của các hợp chất hữu cơ được xử lý theo phương pháp chung sau, (trừ khi có hướng dẫn khác).
Chú thích - Một vài chất có thể phản ứng bất ngờ và nổ dữ dội khi vô cơ hoá với hydro - peroxit. Những biện pháp an toàn thích hợp cần được áp dụng trong mọi lúc.
Chuyển 1,0 g mẫu thử vào một bình thích hợp, cho 5 ml axit sunfuric và một vài hạt thuỷ tinh và vô cơ hoá ở nhiệt độ không quá 120° cho tới khi bắt đầu thành than, tốt nhất là dùng bếp điện trong một tủ hút (fume hood). (Axit sunfuric có thể cho thêm để làm ướt hoàn toàn một số mẫu, nhưng tổng thể tích thêm vào không nên quá 10 ml).
Sau khi mẫu bước đầu được phân huỷ bằng axit, cẩn thận thêm vào từng giọt hydro peroxit 30 % để phản ứng êm dịu và cần đốt nóng lại giữa các giọt. Một ít giọt đầu tiên phải được cho vào rất chậm và trộn kỹ để tránh phản ứng nhanh, và phải ngừng đun nóng nếu có quá nhiều bọt. Lắc xoáy dung dịch trong bình, để tránh chất không được phản ứng đóng cháy trên thành hoặc đáy của bình trong khi vô cơ hoá. Thêm ít một dung dịch peroxit khi dung dịch bắt đầu sẫm màu và tiếp tục vô cơ hoá cho tới khi chất hữu cơ bị phá huỷ. Tăng từ từ, nhiệt độ của bếp đốt tới 250° - 300° cho tới khi khói sunfua trioxit được thoát ra nhiều và dung dịch trở nên không màu hoặc chỉ còn lại màu vàng rơm nhạt. Làm nguội, cẩn thận thêm vào 10 ml nước, tiếp tục cho bốc hơi cho tới khói đặc và làm nguội. Chuyển có tính cách định lượng dung dịch này vào một phễu chiết với một ít nước trợ giúp.
13.3 Cách tiến hành
Chuyển dung dịch mẫu, đã được xử lý như chỉ dẫn của trong từng chuyên luận riêng vào một phễu chiết và cho thêm 6 ml amoni xitrat (PbT) và 2 ml hydroxylanmin hydroclorua, nếu không có hướng dẫn khác (dùng 10 ml dung dịch xitrat khi xác định chì trong các muối sắt). Cho 2 giọt phenol đỏ vào phễu chiết, và làm cho dung dịch vừa tới kiềm hoá (màu đỏ) bằng cách thêm amoniac đậm đặc vào. Làm nguội, dung dịch (nếu cần) dưới dòng nước chảy, sau đó cho thêm 2 ml kali cyanua. Ngay lập tức chiết dung dịch với từng phần 5,0 ml thuốc thử để chiết dithizon dùng để chiết. Rút phần chiết vào một phễu chiết khác cho đến khi dung dịch dithizon giữ được màu xanh lá cây của nó. Lắc dung dịch dithizon hỗn hợp trong 30 giây với 20 ml axit nitri loãng (1 : 100), loại bỏ lớp clorofooc, thêm vào dung dịch axit 5,0 ml dithizon chuẩn và 4 ml dung dịch amoni - xyanua và lắc trong 30 giây.
Màu đỏ tía trong dung dịch clorofooc của mẫu là do sự có mặt của dithizon chì phải không được vượt quá so với màu trong bình đối chứng có chứa một thể tịch dung dịch chì chuẩn loãng tương dương với tổng lượng chì đã quy định trong chuyên khảo, và đã được xử lý theo cách giống như mẫu.
14 Phép thử giới hạn thuỷ ngân
14.1 Thiết bị phát hiện thuỷ ngân
Sử dụng bất kỳ thiết bị đo phổ hấp thụ nguyên tử thích hợp nào, được trang bị một bộ ghi phản hồi nhanh và có thể đo được bức xạ được hấp thụ bởi hơi thuỷ ngân ở đường cộng hưởng thuỷ ngân 253,6 nm. Một thiết bị đo hoặc máy đo hơi thuỷ ngân, được trang bị một bộ phận ghi dải biến động, cũng có thể thảo mãn được nhu cầu.
14.2 Thiết bị sinh khí
Thiết bị sinh khí được mô tả theo hình 1, bao gồm một dụng cụ đo lưu lượng (a), có thể đo tốc độ lưu lượng 2,7 l/h, được nối qua một khoá ba cửa (b) có nút Teflon tới các chai rửi khí có dung tích 125 ml (c và d), và nối tiếp với một ống làm khô có bông thuỷ tinh (e) và cuốicùng là một cuvet hấp thụ chất lỏng bằng thạch anh thích hợp (f), kết thúc với một lỗ thông (g).
Chú thích - Buồng hấp thụ sẽ thay đổi chiều dài đường quang học và, tuỳ thuộc vào loại thiết bị phát hiện thuỷ ngân được sử dụng.
Chai c được lắp với một bình cầu (Corning 31770 125 EC hoặc tương đương), và chai này được đánh dấu một vạch chia 60 ml. ống làm khô e được bọc sơ bộ bằng bông thuỷ tinh hoặc magie peclorat. Chai c được dùng cho dung dịch thử, và chai d vẫn để rỗng trong suốt quá trìnhh thử, được dùng để hứng các giọt nước.
Có thể dùng một thiết bị được lắp ráp như mô tả trong hình vẽ dưới đây. Môi trường sinh khí có thể là không khí nén, hoặc khí nitơ nén.
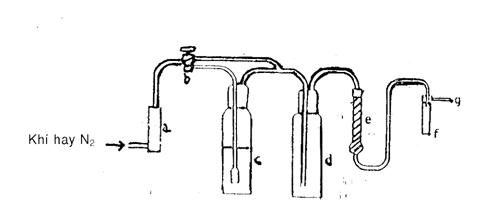
Hình 1 - Thiết bị sinh khí
14.3 Pha dung dịch chuẩn
Cho 1,71 g nitrat thuỷ ngân, Hg(NO3)2.H2O vào một bình định mức dung tích 1000 ml, hoà tan trong một hỗn hợp 100 ml nước và 2 ml axit nitric, pha loãng với nước tới vạch mức và trộn. (Loại bỏ sau một tháng). Cho 10 ml dung dịch này vào bình định mức 1000 ml khác, axit hoá với 5 ml dung dịch axit sunfuric loãng (1: 5), pha loãng với nước tới vạch mức và trộn. (Loại bỏ sau một tuần).
Vào ngày sử dụng, cho 10,0 ml dung dịch thức hai này vào một bình định mức 100 ml, axit hoá với 5 ml dung dịch axit sunfuric loãng (1 : 5), pha loãng với nước tới vạch mức và trộn. Mỗi ml dung dịch này chứa 1 μg Hg. Cho 2,0 ml dung dịch này (2 μg Hg) vào cốc dung tích 50 ml, và thêm vào 20 ml nước, 1 ml dung dịch axit sunfuric loãng (1 : 5), và 1 ml dung dịch thuốc pecmanganat kali (1 : 25). Đậy cốc này lại bằng một nắp kính đồng hồ, đun sôi trong vài giây và để nguội.
14.4 Xử lý mẫu
Xử lý như hướng dẫn trong các chuyên luận riêng.
14.5 Cách tiến hành
Lắp thiết bị sục khí như trong hình 1, với các chai c và d không đựng gì, rỗng và khoá b ở vị trí đường dẫn phụ. Nối thiết bị này với buồng hấp thụ (f) trong thiết bị, điều chỉnh lưu lượng không khí hoặc khí nitơ sao cho, cách tiến hành sau đây, đạt được khả năng tái tạo lắp lại và độ hấp thụ lớn nhất mà không sinh quá nhiều bọt trong dung dịch thử. Thu được số đọc đường nền ở 253,6 nm, phải tuân theo các chỉ dẫn sử dụng thiết bị của nhà sản xuất. Xử lý việc pha dung dịch chuẩn như sau :
Huỷ bỏ thuốc tím thừa bằng cách thêm dung dịch clorua hydroxylamin 1 : 10, nhỏ giọt cho đến khi dung dịch mất màu. Rửa ngay dung dịch này trong chai c bằng nước, và pha loãng với nước tới vạch 60 ml. Thêm 2 ml dung dịch clorua thiếc 10 % (được pha mới mỗi tuần, bằng cách hoà 20 g SnCl22H2O vào 40 ml axit clohydric ấm và pha loãng với 160 ml nước), và nối ngay chai c lại với thiết bị sục khí. Vặn khoá b từ vị trí nhánh sang vị trí sục khí, và ghi nhận số đọc trên thiết bị ghi. Tháo chai c ra khỏi thiết bị sục khí, bỏ hỗn hợp chuẩn, rửa chai c bằng nước, lặp lại trình tự nêu trên với mẫu xử lý; Mọi chất hấp thụ sinh ra trong quá trình xử lý mẫu không được vượt quá lượng chất hấp thụ trong mẫu tiêu chuẩn.
Hoà tan 10 g mẫu với lượng nước đủ để tạo ra 20 ml, thêm 3 ml bromin và 2 ml dung dịch axit xitric 20 % W/V, trộn và thêm 10 ml thuốc thử amoniac và 1 ml thuốc thử dimetylglyxerin. Trộn, pha loãng tới 50 ml bằng nước rồi để yên trong 5 phút; mọi màu sắc sinh ra không được đậm hơn màu sinh ra do cách xử lý tương tự, cùng lúc, 1,0 ml dung dịch niken chuẩn [10 mg/kg Ni được điều chế bằng cách pha loãng 1,0 ml dung dịch clorua niken 0,401 % W/V (NiCl2.2H2O cấp thuốc thử phân tích) với nước thành 100,0 ml] được pha loãng thành 20 ml với nước (0,5 mg/kg Ni).
16.1 Thuốc thử
a) Dung dịch diaminonaphthalen - 2,3
Vào ngày dùng, hoà tan 100 mg 2,3 diaminonaphthalen (C10H10N2) và 500 mg hydroxylamin hydrochlorua (NH2OH.HCl) vào trong axit clohydric đủ để tạo thành 100 ml.
b) Dung dịch selen chuẩn
Cho 120,0 mg selen kim loại dạng bột vào một bình định mức 1000 ml, và hoà tan trong 100 ml axit nitric loãng (1 : 2), làm ấm dần trên thiết bị cách hơi nước để gây tác dụng với dung dịch. Làm mát, pha loãng với nước đến vạch mức và trộn. Cho 5,0 ml dung dịch này vào bình định mức 200 ml, pha loãng với nước tới vạch và trộn. Mỗi ml dung dịch này có chứa 3 μg selen (Se).
16.2 Phương pháp I
16.2.1 Pha dung dịch chuẩn
Cho 2,0 ml dung dịch selen chuẩn vào một cốc dung tích 150 ml, thêm 50 ml axit nitric 0,25 N vào và trộn.
16.2.2 Xử lý mẫu
Cho vào một bình đốt dung tích 1000 ml một lượng mẫu như đã xác định trong chuyên luận riêng (và oxit magie, khi cần), và tiến hành như hướng dẫn trong phần đốt cháy trong bình oxy, dùng axit nitric 0,5 N làm chất lỏng hấp thụ. (Chú ý rằng nếu mẫu có chứa nước thuỷ hoá hoặc độ ẩm cao hơn 1 % thì cho sấy khô ở nhiệt độ 140° trong 2 h trước đốt cháy). Khi kết thúc phản ứng đốt cháy, cho một vài mililit nước vào trong cốc hoặc miệng bình đốt, nới lỏng nút bình, rửa qua nút, vật chứa mẫu, và thành bình bằng khoảng 25 ml nước. Cho dung dịch này vào một cốc có dung tích 150 ml, đun nhỏ lửa cho đến sôi, giữ sôi 10 phút, để nguội.
16.2.3 Cách tiến hành
Dùng hydroxit amoni loãng (1 : 2), điều chỉnh pH của dung dịch chuẩn, của mẫu xử lý, và của 50 ml axit nitric 0,25 N, để làm dung dịch đối chứng, tới giá trị 2,0 ± 0,2. Thêm 200 mg hydroxylanmin hydrochlorua vào mỗi cốc, xoay nhẹ để hoà tan, sau đó thêm ngay 5 ml dung dịch 2,3 diaminonaphthalen vào mỗi dung dịch và trộn. Đậy mỗi bình bằng một nắp kính và để yên ở nhiệt độ phòng 100 phút. Chuyển các dung dịch này vào từng phễu chiết riêng với sự hỗ trợ của khoảng 10 ml nước, chiết xuất mỗi dung dịch bằng 5,0 ml cyclohexan, lắc mạnh từng phễu chiết trong 2 phút, và để yên (cho tách lớp) cho các lớp phân tầng. Loại bỏ các pha nước, và quay li tâm các chất chiết cyclohexan để bỏ hết nước. Xác định độ hấp thu của mỗi chất chiết trong một cuvet 1 cm ở bước sóng tối đa khoảng 380 nm, với một quang phổ thích hợp, sử dụng mẫu trắng để điều chỉnh thiết bị. Độ hấp thu của dịch chiết từ mẫu xử lý (bằng chế phẩm mẫu) không được lớn hơn độ hấp thu từ dung dịch chuẩn khi thử một mẫu cỡ 200 mg, hoặc không lớn hơn (một nửa) 0,5 lần độ hấp thụ của dịch chiết từ dung dịch chuẩn khi thử một mẫu cỡ 100 mg.
16.3 Phương pháp II
16.3.1 Pha dung dịch chuẩn
Cho 0,2 ml dung dịch selen chuẩn vào một cốc dung tích 150 ml, thêm 50 ml axit clohydric 2 N vào, và trộn.
16.3.2 Xử lý mẫu
Cho vào một cốc dung tích 150 ml, một lượng mẫu được xác định trong từng chuyên luận riêng, hoà tan trong 25 ml axit clohydric 4 N, lắc xoay nếu cần, đun từ từ cho đến sôi, và vô cơ hoá trên thiết bị cách hơi nước trong 15 phút. Nhấc ra khỏi thiết bị, thêm 25 ml nước, và để nguội, tới nhiệt độ trong phòng.
16.3.3 Cách tiến hành
Đặt các cốc có chứa các dung dịch chuẩn và mẫu xử lý trong một tủ hút (fume hood). Cẩn thận thêm vào mỗi cốc 5 ml hydroxit amoni vào một cốc thứ ba có chứa 50 ml axit clohydrric 2 N để làm mẫu đối chứng. Để các dung dịch nguội, rồi điều chỉnh pH của chúng tới 2,0 ± 0,2 bằng hydroxit amoni loãng (1 : 2). Tiếp tục theo chỉ dẫn trong trình tự tiến hành của phương pháp I, bắt đầu từ “thêm 200 mg hydroxylamin hydrochlorua... “.
17.1 Thiết bị, dụng cụ
Thiết bị thử nghiệm gồm một bình hình nón, miệng sâu hay khum miệng chén, thành dầy, có dung tích phù hợp cho đốt cháy hoàn toàn mẫu thử để xác định selen. Bình này được lắp một nút thuỷ tinh tròn được nối với một giá đỡ mẫu bao gồm một lưới platin lớn và một mảnh nối lưới platin có kích thước khoảng 1,5 cm x 2 cm. Có thể mua dụng cụ thử nghiệm phù hợp. Như Catalog số 6513 - C20 (dung tích 500 ml) và 6513-C30 (dung tích 1.000 ml) từ Công ty Arthur H. Thomas, P.O Box 779, Philadelphia, Pa. 19105. Hoa Kỳ. Các dụng cụ thử nghiệm tương đương có từ các nguồn khác, hoặc các dụng cụ thích hợp khác tuân theo các nguyên tắc mô tả dưới đây cũng có thể dùng được.
17.2 Cách tiến hành
Lưu ý : Người phân tích cần đeo kính bảo hộ và sử dụng một tấm chắn an toàn, thích hợp ở giữa người và dụng cụ thử nghiệm. các biện pháp an toàn hơn cần được xem xét khi cần thiết, để đảm bảo việc bảo vệ người phân tích ở mức cao nhất. Hơn nữa, bình thử phải được làm sạch hết sức cẩn thận và hạn chế không có vết dung môi hữu cơ nào. Cần sấy khô các mẫu chứa nước, hydrat hoá, hay có độ ẩm cao hơn 1 %, ở nhiệt độ 140° trong 2 giờ trước khi đốt.
Cân chính xác khối lượng mẫu đã xác định theo các chuyên luận cụ thể. Các mẫu rắn cần được cân trên một miếng giấy lọc không có halogenua hình vuông cạnh 4 cm, giấy này cần được gấp xung quanh mẫu. Các mẫu chất lỏng có dung lượng không vượt quá 0,2 ml cần được cần trong ống nhỏ xenlulo acetat đã biết khối lượng [có trong Catalog No. 6513-C80 (100 túi) và 6513-C82 (1,000 túi) của công ty Arthur H. Thomas]; các túi gelatin là thích hợp với mẫu chất lỏng có thể tích lớn hơn 0,2 ml.
Chú thích - Các túi gelatin có thể chứa một lượng đáng kể halogenua hay sunfat kết hợp, trong trường hợp này, cần tiến hành thử nghiệm mãu trắng khi cần thiết.
Đặt mẫu cùng với một dây mồi bằng giấy lọc lên trên lưới platin giữ mẫu. Cho chất lỏng hấp thụ vào trong bình, như xác định trong từng chuyên luận hay trong phép thử tổng quát, làm ẩm chỗ nối của nút bằng nước, và đẩy không khí ra khỏi bình bằng cách cho một dòng oxy chảy nhanh vào bình, lắc xoáy chất lỏng để giúp nó hấp thụ oxy.
Chú thích - Bão hoà chất lỏng bằng oxy chủ yếu giúp cho việc thành công của thử nghiệm này.
Đốt dây mồi bằng phương tiện thích hợp. Nếu dải giấy mồi cháy ngoài bình, ngay lập tức phải đẩy nhanh bộ phận giữ mẫu vào trong bình, dốc ngược bình sao cho dung dịch hấp thự tạo ra một lớp bít kín quanh nút, và giữ chặt nút ở đúng vị trí. Nếu việc cháy xảy ra trong hệ thống kín, việc dốc ngược bình có thể bỏ qua. Sau khi hoàn tất việc đốt, lắc mạnh bình và để bình yên không ít hơn 10 phút, thỉnh thoảng lại lắc. Sau đó tiếp túc làm như đã hướng dẫn trong các chuyên luận cụ thể.
(Phương pháp kendal)
Cảnh báo : Cân dẩm bảo thoáng khí trong phòng thí nghiệm và không cho phép tích tụ thuỷ ngân ra ngoài.
Chú thích - Mọi thuốc thử phải không có nitơ, nếu có sẵn, hoặc thuốc thử phải có hàm lượng nitơ rất thấp.
18.1 Phương pháp I
Phương pháp này cần được sử dụng trừ khi có những chỉ dẫn khác trong từng chuyên luận cụ thể. Nó không áp dụng được một vài hợp chất chứa nitơ mà các chất này không cho thấy rằng hàm lượng nitơ toàn bộ, dựa trên việc vô cơ hoá với axit sunfuric.
18.1.1 Không có nitric và nitrat
Trừ khi có những chỉ dẫn khác nếu không thì chuyển khoảng 1 g mẫu, đã được cân chính xác, vào một bình kendal thuỷ tinh cứng 500 ml, gói bọc mẫu, nếu là chất rắn hay nửa rắn, trong giấy lọc không chứa nitơ để tạo điều kiện cho việc chuyển mẫu nếu cần. Cho vào bình này 10 g kali sunfat dạng bột hay natri sunfat khan, 500 mg đồng sunfat dạng bột và 20 ml axit sunfuric. Đun nóng từ từ hỗn hợp này, giữ bình nghiêng một góc khoảng 45°, và sau khi hiện tượng nổi bọt đã ngừng, nhanh chóng đun sôi cho đến khi dung dịch còn lại mầu xanh lá cây sáng hoặc hầu như không có mầu trong 30 phút.
Để nguội và thêm 150 ml nước, trộn lẫn, và lại để nguội. Cẩn thận rót 100 ml dung dịch natri hydroxit (2 : 5) vào bên trong bình sao cho nó tạo nên một lớp dưới lớp dung dịch axit, sau đó cho thêm một vài hạt kẽm. Nối bình với thiết bị chưng cất bao gồm một bình cầu nối với bình kendal và bình ngưng tụ, ống thoát đặt dưới bề mặt của 50 ml dung dịch axit boric (1 : 25) được chứa trong một bình hoặc lọ 500 ml. Xoay nhẹ các chất trong bình kendal để trộn, và chưng cất cho đến khi thu nhận được khoảng 2/3 dung dịch trong bình hứng. Cho thuốc thử methyl đỏ / methylen xanh vào bình này và chuẩn độ với axit sunfuric 0,5 N. Tiến hành thử nghiệm đối chứng bằng cách thay mẫu 2 g sucroz (đường) và làm những hiệu chỉnh cần thiết. Mỗi một ml axit 0,5 N tương đương với 7,003 mg nitơ.
Chú thích - Nếu biết rằng chất thử nghiệm có hàm lượng nitơ thấp, có thể dùng axit 0,1 N thay cho dung dịch 0,5 N, trong trường hợp đó mỗi một ml axit tương đương với 1,401 mg nitơ.
18.1.2 Có mặt nitric và nitrat
Chuyển vào một bình thuỷ tinh cứng kendal 500 ml, một lượng mẫu, đã được cân chính xác, đại diện cho khoảng 150 mg nitơ, thêm 25 ml axit sunfuric trong đó có 1 g axit salicylic đã được hoà tan, trộn, và để yên trong 30 phút, thỉnh thoảng lắc. Cho 5 g natri thiosunfat dạng bột, trộn kỹ, rồi thêm 500 mg đồng sunfat dạng bột hay oxit thuỷ ngân, và tiếp tục như đã hướng dẫn ở mục A, bắt đầu với “đun nóng từ từ hỗn hợp này... “. Trước khi vô cơ hoá các chất được biết có hàm lượng nitơ cao hơn 10 %, cho thêm 500 mg đến 1 g axit benzoic để tạo điều kiện thuận lợi cho việc phân huỷ.
18.2 Phương pháp II (bán vi lượng hay trung lượng)
Chuyển một lượng mẫu đã được cân hay đo chính xác, tương đương với khoảng 2 hay 3 mg nitơ vào trong bình vô cơ hoá của thiết bị kendal bán vi lượng. Cho thêm 1 g hỗn hợp bột kali sunfat và đồng sunfat (10 : 1), dùng một tia nước nhỏ để rửa bất kỳ vật liệu nào dính bám vào miệng bình xuống, sau đó rót 7 ml axit sunfuric vào trong thành bình để tráng rửa nó. Cẩn thận thêm vào bình 1 ml hydrogen peroxid 30 %, lắc tròn bình trong khi cho các chất này.
(Cảnh báo : Không được thêm bất kỳ lượng peroxid nào trong khi vô cơ hoá). Nung nóng trên ngọn lửa hay bếp điện cho đến khi dung dịch đã có màu xanh rõ và thành bình không còn vệt than hoá. Cẩn thận thêm 20 ml nước, để nguội, sau đó cho qua phễu 30 ml dung dịch hydroxid natri (2 : 5) và tráng rửa phễu bằng 10 ml nước. Nối bình với thiết bị chưng cất hơi và ngay lập tức bắt đầu việc chưng cất bằng hơi. Thu lấy thành phẩm chưng cất trong 15 ml dung dịch axit boric (1 : 25), dung dịch này đã được cho 3 giọt thuốc thử methyl đỏ / methylen xanh và đủ nước để bao trùm miệng thoát ống ngưng tụ. Tiếp tục chạy hơi nước cho đến khi thu được 80 - 100 ml thành phẩm chưng cất, sau đó tháo bình hấp thụ, tráng rửa miệng thoát ngưng bằng một ít nước, và đem chuẩn độ với axit sunfuric 0,01 N. Mỗi một ml axit 0,01 N tương đương với 0.140 mg (140 μg) nitơ.
Khi có nhiều hơn 2 hay 3 mg nitơ trong lượng mẫu được cân để thửn ghiệm, có thể dùng axit sunfuric 0,02 hay 0,1 N để chuẩn độ, thì cần ít nhất 15 ml chất chuẩn độ. Nếu khối lượng khô tổng cộng của sản phẩm lớn hơn 100 mg thì sẽ tăng theo tỷ lệ lượng axit sunfuric và natri hydroxit được cho vào trước khi chưng cất.
Trừ khi có các chỉ dẫn khác, nếu không thì chuyển 100 ml mẫu vào một chén dùng để bay hơi bằng platin đã cân bì dung tích 125 ml, đã được sấy nóng từ trước ở 105° tới khối lượng không đổi, và cho bốc hơi mẫu tới khô trên bếp cánh hơi. Sấy nóng ở nhiệt độ 105°C, trong 30 phút hoặc tới khối lượng không đổi, để nguội trong một bình hút ẩm và cân.
Trừ khi có các chỉ dẫn khác, nếu không thì cho một lượng mẫu đã xác định vào một ống Nessler, hoà tan nó trong khoảng 30 ml nước và trung hoà bằng thuốc thử axit clohydric loãng nếu dung dịch là kiềm. Thêm 1 ml thuốc thử axit clohydric loãng và dùng nước pha loãng tới 50 ml. Nếu sử dụng mẫu ở dạng dung dịch, chuyển dung dịch mẫu này vào một ống Nessler và dùng nước pha loãng tới 50 ml. Chuyển một lượng axit sunfuric 0,01 N như trên vào một ống Nessler khác để dùng làm chuẩn, thêm 1 ml thuốc thử axit clohydric loãng, và pha loãng với nước tới 50 ml.
Nếu dung dịch chứa mẫu không được trong, phải lọc cả hai dung dịch này trong cùng một điều kiện. Cho 2 ml thuốc thử bari clorua vào từng dung dịch, trộn kỹ, và để yên trong 10 phút. So sánh độ đục của hai dung dịch bằng cách quan sát các ống Nessler từ bên thành ống và từ đỉnh đặt dung dịchối diện với một nền đen.
Độ đục của mẫu không được vượt quá độ đục chuẩn.
(Phương pháp chuẩn độ Karl Fischer)
21.1 Nguyên tắc
Phương pháp chuẩn độ Karl Fischer để xác định nước được dựa trên phản ứng định lượng của nước với một dung dịch khan của sunfua dioxit và iôt được hoà tan trong pyridin và một chất cồn (alcohol).
Mẫu có thể được chuẩn độ trực tiếp với thuốc thử, hoặc nhà phân tích có thể tiến hành một phương pháp chuẩn độ dư. Trong phép chuẩn độ dư, thuốc thử ở mức dư được thêm vào mẫu, phải để đủ thời gia cho phản ứng đạt tới hoàn toàn, và thuốc thử không sử dụng hết được chuẩn với dung dịch nước chuẩn trong methnol. Phương pháp chuẩn độ dư nói chung có thể được áp dụng được và tránh những khó khăn có thể xuất hiện trong chuẩn độ trực tiếp tung đó các chất có nước kết hợp thoát ra chậm.
Phép tính hệ số tỷ lượng (Stoichiometry) của phản ứng thì không chính xác, và độ tái lặp của một lần thử phụ thuộc vào các yếu tố như nồng độ tương đối của các thành phần thuốc thử, bản chất của dung môi trơ được dùng để hoà tan mẫu, và kỹ thuật được dùng trong phép xác định riêng biệt. Vì thế, cần phải dùng một kỹ thuật đã được chuẩn hoá theo kinh nghiệm nhằm đạt tới độ chính xác mong muốn.
Độ chính xác của phương pháp thu được bởi độ mở của phương pháp, trong đó độ ẩm không khí được loại ra khỏi hệ thống. Việc chuẩn độ nước thông thường được thực hiện với việc dùng methnol khan làm dung môi cho mẫu; tuy nhiên, có thể dùng các dung môi thích hợp khác đối với các chất đặc biệt hoặc không phổ biến lắm.
21.1 Thiết bị và phép xác định điểm cuối cùng
Có thể dùng bất kỳ dụng cụ thử nghiệm nào có thể loại bỏ thích đáng độ ẩm không khí và xác định được điểm cuối cùng. Trong trường hợp dung dịch không mầu, thì chuẩn độ trực tiếp, điểm cuối cùng có thể quan sát bằng mắt khi thay đổi mầu từ vàng hoàng yến sang mầu hổ phách. Trường hợp ngược lại được quan sát khi mẫu được chuẩn độ dư. Tuy nhiên, phổ biến hơn, điểm cuối cùng được xác định theo phép đo điện tử với việc sử dụng các điện cực platin đối ngẫu (khoảng 5 mm vuông và cách nhau 2,5 cm) và một dòng phân cực khoảng 100 àA ở điện thế sử dụng khoảng 200 mV. Khi kết thúc phản ứng, một sự thay đổi trong tính chất điện hoá của dung dịch sẽ được cảm nhận bởi các điện cực và điểm cuối cùng được chỉ báo bởi độ lệch của microampe kế hay bằng một vài thiết bị cảm ứng dòng hay thiết bị cảm ứng điện thế. Với một vài thiết bị chuẩn độ tự động, sự thay đổi đột ngột về dùng hay điện thế tại điểm cuối cùng sẽ làm đóng van solenoid, van này kiểm soát buret phân phối chất chuẩn độ. Các thiết bị hiện có trên thị trường nói chung bao gồm một hệ thống kín gồm một hay hai buret tự động và một bình chuẩn độ đậy chặt, được lắp với các điện cực thích hợp và một máy khuấy từ. Không khí trong hệ thống được giữ khô với chất làm khô thích hợp như photpho pentoxid và bình chuẩn độ có thể được làm sạch bởi một dòng nitơ khô hay dòng không khí khô.
21.3 Pha thuốc thử Fischer
Thêm 125 g iốt vào một bình chứa hỗn hợp gồm 670 ml methanol và 170 ml pyridin, ngay sau đó đậy nút bình, và làm lạnh. Cho chạy sunfua dioxit khô qua 100 ml pyridin được chứa trong một bình khắc độ 250 ml và được làm lạnh trong một chậu đá cho đến khi thể tích của dung dịch đạt được bằng 200 ml. Cho từ từ dung dịch này, đồng thời lắc đều, vào hỗn hợp iốt lạnh, đậy nắp ngay, và lắc kỹ cho đến khi iốt hoà tan. Chuyển dung dịch kết hợp này sang thiết bị thử nghiệm, tốt hơn cả là một buret tự động được bảo vệ tránh ẩm nhờ các chất chống ẩm như photpho pentoxid, calci chlorua khan, hay gel silica, và để yên trong 24 giờ hay qua đêm trước khi chuẩn hoá. Mỗi một ml của thuốc thử này khi mới pha tương đương với khoảng 5 mg nước. Do dung dịch này liên tục suy giảm nó cần được chuẩn hoá trong vòng 1 giờ trước khi dùng, hay hàng ngày nếu sử dụng liên tục. Bảo vệ tránh bị ánh sáng mặt trời trong khi dùng, và lưu giữ các dung dịch gốc trong bình có nút thuỷ tinh và trong tủ lạnh.
Thuốc thử Karl Fischer ổn định hoá có bán trên thị trường có thể đáp ứng yêu cầu thay thế cho dung dịch đã chỉ dẫn ở đây.
21.4 Chuẩn hoá thuốc thử Fischer
21.4.1 Chuẩn hoá ban đầu
Cho 35 tới 40 ml methanol vào bình chuẩn độ của thiết bị thử nghiệm và chuẩn độ với thuốc thử tới mầu điểm cuối hay tới điểm cuối của phép đo điện.
a) Để xác định lượng vết của nước (tức là, ít hơn khoảng 1 % trong mẫu thử), có thể dung natri tartrat dihydrat (Na2C4H4O6.2H2O), được cân chính xác, vào methanol, và lại chuẩn độ tơi điểm cuối với thuốc thử này.
Tính toán hệ số tương đương nước, F, tính bằng mg trên ml của chất thử bằng công thức :
2 x (18.02 / 230.08) x (W/V)
Trong đó
W là khối lượng natri tartrat dihydrat, tính bằng mg;
V là thể tích thuốc thử tiêu thụ trong lần chuẩn độ thứ hai, tính bằng ml;
18,02 và 230,08 tương ứng là khối lượng phân tử của nước và natri tartrat dihydrat.
b) Trong phép thử chính xác đối với lượng nước nhiều hơn vết (tức là hơn khoảng 1 %), dùng nước cất như là chất tham chiếu. Nếu dùng phương pháp này, nhanh chóng thêm 25 tới 250 mg nước cất đã được cân chính xác, vào methanol, và lại chuẩn độ tới điểm cuối với thuốc thử.
Tính hệ số tương đương nước, F, bằng số mg nước trên ml của chất thử, theo công thức W/V
Trong đó
W là khối lượng nước cất, tính bằng mg;
V là thể tích thuốc thử tiêu tốan trong chuẩn độ lần thứ hai, tính bằng ml.
21.4.2 Chuẩn hoá lần hai
Pha một dung dịch nước methanol bằng cách pha loãng 2 ml nước với methanol thành 1000 ml. Chuẩn hoá dung dịch này bằng chuẩn độ 25,0 ml với thuốc thử, đã được chuẩn hoá từ trước như đã chỉ dẫn trong mục chuẩn hoá ban đầu ở trên. Tính toán hàm lượng nước, bằng mg trên ml, của dung dịch nước methanol theo công thức :
V’F / 25
Trong đó :
V’ là thể tích thuốc thử đã bị tiêu tốn, tính bằng ml;
F là hệ số tương đương nước của thuốc thử đã được xác định như đã chỉ dẫn trong “chuẩn hoá ban đầu“. Hàm lượng nước của dung dịch nước methanol cần được xác định hàng tuần và thuốc thử được chuẩn hoá với nó cũng xác định theo định kỳ, nếu cần.
21.5 Cách tiến hành
Chú thích - Cần xác định hàm lượng nước bằng phương pháp chuẩn độ trực tiếp, trừ khi có các chỉ dẫn khác.
21.5.1 Chuẩn độ trực tiếp
Ngoại trừ có các chỉ dẫn khác, nếu không thì đặt khoảng 35 đến 40 ml methanol vào bình chuẩn độ và chuẩn độ với thuốc thử tới điểm cuối, không cần để ý đến lượng đã tiêu tốn. Nhanh chóng chuyển sang bình chuẩn độ một lượng mẫu đã được cân hay đo chính xác, tốt nhất có chứa 10 đến 50 mg nước, khuấy mạnh, và chuẩn độ lại tới điểm cuối. Hàm lượng nước của mẫu, tính bằng mg, nhận được bằng cách nhận lượng thuốc thử đã dùng trong chuẩn độ mẫu với hệ số tương đương F của thuốc thử.
21.5.2 Chuẩn độ dư
Ngoại trừ có các chỉ dẫn khác, nếu không thì khoảng 35 đến 40 ml methanol vào bình chuẩn độ và chuẩn độ với thuốc thử tới điểm cuối, không cần để ý đến lượng đã tiêu thụ. Nhanh chóng chuyển sang bình chuẩn độ một lượng mẫu đã được cân hay đo chính xác, tốt nhất có chứa 10 đến 50 mg nước, khuấy mạnh, và thêm một lượng dư được cân hay đo chính xác của thuốc thử. Để yên đủ thời gian cho phan ứng đạt tới mức hoàn toàn và chuẩn độ thuốc thử không tiêu thụ hết với dung dịch nước methanol đã chuẩn hoá tới điểm cuối.
Hàm lượng nước trong mẫu, tính bằng mg, sẽ nhận được bằng cách nhân lượng thuốc thử đã dùng trong chuẩn độ mẫu với hệ số tương đương F của thuốc thử.